http://www.kidney-international.org
KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease
KDIGO gratefully acknowledges the following consortium of sponsors that make our initiatives possible: Abbott, Amgen, Bayer Schering Pharma, Belo Foundation, Bristol-Myers Squibb, Chugai Pharmaceutical, Coca-Cola Company, Dole Food Company, Fresenius Medical Care, Genzyme, Hoffmann-LaRoche, JC Penney, Kyowa Hakko Kirin, NATCO - The Organization for Transplant Professionals, NKF - Board of Directors, Novartis, Pharmacosmos, PUMC Pharmaceutical, Robert and Jane Cizik Foundation, Shire, Takeda Pharmaceutical, Transwestern Commercial Services, Vifor Pharma, and Wyeth.
Sponsorship Statement: KDIGO is supported by a consortium of sponsors and no funding is accepted for the development of specific guidelines.
KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease
Tables and Figures
KDIGO Board Members
Reference Keys
Abbreviations and Acronyms
Notice
Foreword
Work Group Membership
Abstract
Summary of Recommendation Statements
Chapter 1: Diagnosis and evaluation of anemia in CKD
Chapter 2: Use of iron to treat anemia in CKD
Chapter 3: Use of ESAs and other agents to treat anemia in CKD
Chapter 4: Red cell transfusion to treat anemia in CKD
References
TABLES
| Table 1. | Hb levels in children between 1-19 years for initiation of anemia workup |
| Table 2. | Hb levels in children between birth and 24 months for initiation of anemia workup |
| Table 3. | Potentially correctable versus non correctable factors involved in the anemia of CKD, in addition to ESA deficiency |
| Table 4. | Practical approach in presence of ESA hyporesponsiveness |
| Table 5. | Estimated risk associated with blood transfusions per unit transfused |
| Table 6. | Estimated risk of transfusion-related infections per unit transfused |
| Table 7. | Indications for blood transfusions |
| Table 8. | Systematic review topics and screening criteria |
| Table 9. | Hierarchy of importance of outcomes |
| Table 10. | Literature search yield of primary articles for systematic review topics |
| Table 11. | Classification of study quality |
| Table 12. | GRADE system for grading quality of evidence |
| Table 13. | Final grade for overall quality of evidence |
| Table 14. | Balance of benefits and harm |
| Table 15. | KDIGO nomenclature and description for grading recommendations |
| Table 16. | Determinants of strength of recommendation |
| Table 17. | The Conference on Guideline Standardization (COGS) checklist for reporting clinical practice guidelines |
FIGURES
| Figure 1. | Receiver operating characteristic (ROC) curves, examining the utility of iron status tests to distinguish iron deficient from nondeficient study patients |
| Figure 2. | Sensitivity and specificity of TSAT and serum ferritin and their combination (TSAT + ferritin) and bone marrow iron (BM iron) to identify correctly a positive erythropoietic response (Z1-g/dl [Z10-g/l] increase in Hb [DHb]) to intravenous iron in 100 nondialysis patients with CKD (areas under the ROCs) |
| Figure 3. | Lymphocytotoxic antibody reactivity against random donor test panel in relation to the number of blood transfusions |
| Figure 4. | Algorithms for red cell transfusion use in CKD patients |
Additional information in the form of supplementary materials can be found online at http://www.kdigo.org/clinical_practice_guidelines/anemia.php
KDIGO Board Members
|
Norbert Lameire, MD, PhD Founding KDIGO Co-Chairs
Immediate Past Co-Chair |
|
|
Bertram L Kasiske, MD
Omar I Abboud, MD, FRCP |
David C Wheeler, MD, FRCP
Michel Jadoul, MD |
NKF-KDIGO GUIDELINE DEVELOPMENT STAFF
Kerry Willis, PhD, Senior Vice-President for Scientific Activities
Michael Cheung, MA, Guideline Development Director
Sean Slifer, BA, Guideline Development Manager
Reference Keys
NOMENCLATURE AND DESCRIPTION FOR RATING GUIDELINE RECOMMENDATIONS
Within each recommendation, the strength of recommendation is indicated as Level 1, Level 2, or Not Graded, and the quality of the supporting evidence is shown as A, B, C, or D.
| Grade* | Implications | ||
|---|---|---|---|
| Patients | Physicians | Policy | |
| Level 1 'We recommend' |
Most people in your situation would want the recommended course of action and only a small proportion would not. | Most patients should receive the recommended course of action. | The recommendation can be evaluated as a candidate for developing a policy or a performance measure. |
| Level 2 'We suggest' |
The majority of people in your situation would want the recommended course of action, but many would not. | Different choices will be appropriate for different patients. Each patient needs help to arrive at a management decision consistent with her or his values and preferences. | The recommendation is likely to require substantial debate and involvement of stakeholders before policy can be determined. |
*The additional category 'Not Graded' was used, typically, to provide guidance based on common sense or where the topic does not allow adequate application of evidence. The most common examples include recommendations regarding monitoring intervals, counseling, and referral to other clinical specialists. The ungraded recommendations are generally written as simple declarative statements, but are not meant to be interpreted as being stronger recommendations than Level 1 or 2 recommendations.
| Grade | Quality of evidence | Meaning |
|---|---|---|
| A | High | We are confident that the true effect lies close to that of the estimate of the effect. |
| B | Moderate | The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. |
| C | Low | The true effect may be substantially different from the estimate of the effect. |
| D | Very Low | The estimate of effect is very uncertain, and often will be far from the truth. |
STAGES OF CHRONIC KIDNEY DISEASE
| CKD Stage | Description | GFR (ml/min per 1.73 m2) |
|---|---|---|
| 1 | Kidney damage with normal or increased GFR | ≥ 90 |
| 2 | Kidney damage with mild decreased GFR | 60-89 |
| 3 | Moderate decreased GFR | 30-59 |
| 4 | Severe decreased GFR | 15-29 |
| 5a | Kidney failure | <15 (or dialysis) |
CKD, chronic kidney disease; GFR, glomerular filtration rate.
CKD 1-5T notation applies to kidney transplant recipients.
a5D if dialysis (HD or PD).
CURRENT CHRONIC KIDNEY DISEASE (CKD) NOMENCLATURE USED BY KDIGO
| CKD Stage | Definition |
|---|---|
| CKD | CKD of any stage (1-5), with or without a kidney transplant, including both non-dialysis dependent CKD (CKD 1-5ND) and dialysis-dependent CKD (CKD 5D) |
| CKD ND | Non-dialysis-dependent CKD of any stage (1-5), with or without a kidney transplant (i.e., CKD excluding CKD 5D) |
| CKD T | Non-dialysis-dependent CKD of any stage (1-5) with a kidney transplant |
| Specific CKD Stages | |
| CKD 1, 2, 3, 4 | Specific stages of CKD, CKD ND, or CKD T |
| CKD 3-4, etc. | Range of specific stages (e.g., both CKD 3 and CKD 4) |
| CKD 5D | Dialysis-dependent CKD 5 |
| CKD 5HD | Hemodialysis-dependent CKD 5 |
| CKD 5PD | Peritoneal dialysis-dependent CKD 5 |
CONVERSION FACTORS OF METRIC UNITS TO SI UNITS
| Parameter | Metric units | Conversion factor | SI units |
|---|---|---|---|
| Ferritin | ng/ml | 1 | µg/l |
| Hemoglobin | g/dl | 10 | g/l |
Abbreviations and Acronyms
| Δ | Change |
| AGREE | Appraisal of Guidelines for Research and Evaluation |
| BM | Bone marrow |
| CBC | Complete blood count |
| CERA | Continuous erythropoietin receptor activator |
| CHOIR | Correction of Hemoglobin and Outcomes in Renal Insufficiency |
| CI | Confidence interval |
| CKD | Chronic kidney disease |
| CKiD | Chronic Kidney Disease in Children Prospective Cohort Study |
| COGS | Conference on Guideline Standardization |
| CREATE | Cardiovascular Risk Reduction by Early Anemia Treatment With Epoetin Beta Trial |
| CRP | C-reactive protein |
| CVD | Cardiovascular disease |
| eGFR | Estimated glomerular filtration rate |
| EMA | European Medicines Agency |
| EPO | Erythropoietin |
| ERT | Evidence review team |
| ESA | Erythropoiesis-stimulating agent |
| ESRD | End-stage renal disease |
| EQ-5D | A measure of health status from the EuroQol Group |
| FACT-Fatigue | Functional Assessment of Cancer Therapy-Fatigue |
| FDA | Food and Drug Administration |
| GFR | Glomerular filtration rate |
| GRADE | Grading of Recommendations Assessment, Development, and Evaluation |
| Hb | Hemoglobin |
| Hct | Hematocrit |
| HCV | Hepatitis C virus |
| HD | Hemodialysis |
| HEMO Study | Kidney Disease Clinical Studies Initiative Hemodialysis Study |
| HLA | Human leukocyte antigen |
| HR | Hazard ratio |
| IM | Intramuscular |
| IU | International unit |
| IV | Intravenous |
| KDIGO | Kidney Disease: Improving Global Outcomes |
| KDOQI | Kidney Disease Outcomes Quality Initiative |
| Kt/V | Clearance expressed as a fraction of urea or body water volume |
| MCH | Mean corpuscular hemoglobin |
| NAPRTCS | North American Pediatric Renal Transplant Cooperative Study |
| ND | Non-dialysis |
| NHANES | National Health and Nutrition Examination Survey |
| PD | Peritoneal dialysis |
| PRA | Panel reactive antibody |
| PRCA | Pure red cell aplasia |
| QoL | Quality of life |
| RBC | Red blood cell |
| RCT | Randomized controlled trial |
| rHuEPO | Recombinant human erythropoietin |
| ROC | Receiver operating characteristic |
| RR | Relative risk |
| SC | Subcutaneous |
| SF-36 | 36-Item Medical Outcomes Study Short-Form Health Survey |
| TRALI | Transfusion-related acute lung injury |
| TREAT | Trial to Reduce Cardiovascular Events with Aranesp Therapy |
| TSAT | Transferrin saturation |
| USRDS | United States Renal Data System |
| WHO | World Health Organization |
Notice
SECTION I: USE OF THE CLINICAL PRACTICE GUIDELINE
This Clinical Practice Guideline document is based upon systematic literature searches last conducted in October 2010, supplemented with additional evidence through March 2012. It is designed to provide information and assist decision making. It is not intended to define a standard of care, and should not be construed as one, nor should it be interpreted as prescribing an exclusive course of management. Variations in practice will inevitably and appropriately occur when clinicians take into account the needs of individual patients, available resources, and limitations unique to an institution or type of practice. Every health-care professional making use of these recommendations is responsible for evaluating the appropriateness of applying them in any particular clinical situation. The recommendations for research contained within this document are general and do not imply a specific protocol.
SECTION II: DISCLOSURE
Kidney Disease: Improving Global Outcomes (KDIGO) makes every effort to avoid any actual or reasonably perceived conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the Work Group. All members of the Work Group are required to complete, sign, and submit a disclosure and attestation form showing all such relationships that might be perceived or actual conflicts of interest. This document is updated annually and information is adjusted accordingly. All reported information will be printed in the final publication and are on file at the National Kidney Foundation (NKF), Managing Agent for KDIGO.
Copyright © 2012 by KDIGO. All rights reserved.
Single photocopies may be made for personal use as allowed by national copyright laws.
Special rates are available for educational institutions that wish to make photocopies for
non-profit educational use. No part of this publication may be reproduced, amended, or
transmitted in any form or by any means, electronic or mechanical, including photocopying,
recording, or any information storage and retrieval system, without explicit permission in
writing from KDIGO. Details on how to seek permission for reproduction or translation,
and further information about KDIGO's permissions policies can be obtained by contacting
Anita Viliusis, KDIGO Permissions Manager, at [email protected]
To the fullest extent of the law, neither KDIGO, Kidney International Supplements, National
Kidney Foundation (KDIGO Managing Agent) nor the authors, contributors, or editors,
assume any liability for any injury and/or damage to persons or property as a matter of
products liability, negligence or otherwise, or from any use or operation of any methods,
products, instructions, or ideas contained in the material herein.
Foreword
It is our hope that this document will serve several useful purposes. Our primary goal is to improve patient care. We hope to accomplish this, in the short term, by helping clinicians know and better understand the evidence (or lack of evidence) that determines current practice. By providing comprehensive evidence-based recommendations, this guideline will also help define areas where evidence is lacking and research is needed. Helping to define a research agenda is an often neglected, but very important, function of clinical practice guideline development.
We used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) system to rate the strength of evidence and the strength of recommendations. In all, there were only 2 (5.4%) recommendations in this guideline for which the overall quality of evidence was graded 'A,' whereas 9 (24.3%) were graded 'B,' 14 (37.8%) were graded 'C,' and 12 (32.4%) were graded 'D.' Although there are reasons other than quality of evidence to make a grade 1 or 2 recommendation, in general, there is a correlation between the quality of overall evidence and the strength of the recommendation. Thus, there were 15 (40.5%) recommendations graded '1' and 22 (59.5%) graded '2.' There were 2 (5.4%) recommendations graded '1A,' 8 (21.6%) were '1B,' 1 (2.7%) were '1C,' and 4 (10.8%) were '1D.' There were 0 (0%) graded '2A,' 1 (2.7%) were '2B,' 13 (35.1%) were '2C,' and 8 (21.6%) were '2D.' There were 22 (37.3%) statements that were not graded.
Some argue that recommendations should not be made when evidence is weak. However, clinicians still need to make clinical decisions in their daily practice, and they often ask, 'What do the experts do in this setting?' We opted to give guidance, rather than remain silent. These recommendations are often rated with a low strength of recommendation and a low strength of evidence, or were not graded. It is important for the users of this guideline to be cognizant of this (see Notice). In every case these recommendations are meant to be a place for clinicians to start, not stop, their inquiries into specific management questions pertinent to the patients they see in daily practice.
We wish to thank the Work Group Co-Chairs, Drs John McMurray and Pat Parfrey, along with all of the Work Group members who volunteered countless hours of their time developing this guideline. We also thank the Evidence Review Team members and staff of the National Kidney Foundation who made this project possible. Finally, we owe a special debt of gratitude to the many KDIGO Board members and individuals who volunteered time reviewing the guideline, and making very helpful suggestions.
Bertram L Kasiske, MD
KDIGO Co-Chair
David C Wheeler, MD, FRCP
KDIGO Co-Chair
Work Group Membership
| WORK GROUP CO-CHAIRS | |
|---|---|
| John J V McMurray, MD, FRCP, FESC BHF Glasgow Cardiovascular Research Centre Glasgow, United Kingdom |
Patrick S Parfrey, MD, FRCPC, FRSC Memorial University Medical School St John's, Canada |
| WORK GROUP | |
|
John W Adamson, MD University of California at San Diego San Diego, CA, USA Pedro Aljama, MD, PhD Hospital Universitario Reina Sofia Córdoba, Spain Jeffrey S Berns, MD The Perelman School of Medicine at the University of Pennsylvania Philadelphia, PA, USA Julia Bohlius, MD, MScPH University of Bern Bern, Switzerland Tilman B Drüeke, MD, FRCP Université de Picardie Jules Verne Amiens, France Fredric O Finkelstein, MD Yale University New Haven, CT, USA Steven Fishbane, MD North Shore-LIJ Health System Manhasset, NY, USA Tomas Ganz, PhD, MD David Geffen School of Medicine at UCLA Los Angeles, CA, USA |
Iain C Macdougall, BSc, MD, FRCP King's College Hospital London, United Kingdom Ruth A McDonald, MD Seattle Children's Hospital Seattle, WA, USA Lawrence P McMahon, MBBS, MD Monash University Box Hill, Australia Gregorio T Obrador, MD, MPH Universidad Panamericana School of Medicine Mexico City, Mexico Giovanni FM Strippoli, MD, PhD, MPH Consorzio Mario Negri Sud Chieti, Italy Günter Weiss, MD Medical University of Innsbruck Innsbruck, Austria Andrzej Wieçek, MD, PhD, FRCP Silesian University School of Medicine Katowice, Poland |
| EVIDENCE REVIEW TEAM | |
|
Tufts Medical Center, Boston, MA, USA: Ethan M Balk, MD, MPH; Project Director; Program Director, Evidence-based Medicine Ashish Upadhyay, MD, Assistant Project Director Dana C Miskulin, MD, MS, Staff Nephrologist Amy Earley, BS, Project Coordinator Shana Haynes, MS, DHSc, Research Assistant Jenny Lamont, MS, Project Manager In addition, support and supervision were provided by: Katrin Uhlig, MD, MS; Director, Guideline Development |
|
Abstract
The 2012 Kidney Disease: Improving Global Outcomes (KDIGO) Clinical Practice Guideline forAnemia in Chronic Kidney Disease aims to provide guidance on diagnosis, evaluation,management and treatment for all CKD patients (non-dialysis, dialysis, kidney transplantrecipients and children) at risk of or with anemia. Guideline development followed an explicitprocess of evidence review and appraisal. The guideline contains chapters addressing diagnosisand evaluation of anemia in CKD and the use of various therapeutic agents (iron, ESAs andother agents) and red cell transfusion as means of treatment. Treatment approaches areaddressed in each chapter and guideline recommendations are based on systematic reviews ofrelevant trials. Appraisal of the quality of the evidence and the strength of recommendationsfollowed the GRADE approach. Ongoing areas of controversies and limitations of the evidenceare discussed and additional suggestions are also provided for future research.
Keywords: anemia in CKD; blood transfusions; clinical practice guideline; erythropoiesis stimulating agent; KDIGO; evidence-based recommendation; iron; systematic review.
In citing this document, the following format should be used: Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney inter., Suppl. 2012; 2: 279-335.
Summary of Recommendation Statements
Chapter 1: Diagnosis and evaluation of anemia in CKD
TESTING FOR ANEMIA
Frequency of testing for anemia
Diagnosis of anemia
Investigation of anemia
Chapter 2: Use of iron to treat anemia in CKD
TREATMENT WITH IRON AGENTS
*Based on patient symptoms and overall clinical goals, including avoidance of transfusion, improvement in anemia-related symptoms, and after exclusion of active infection.
**Consistent with Recommendations #3.4.2 and 3.4.3.
***Based on patient symptoms and overall clinical goals including avoidance of transfusion and improvement in anemia-related symptoms, and after exclusion of active infection and other causes of ESA hyporesponsiveness.
IRON STATUS EVALUATION
CAUTIONS REGARDING IRON THERAPY
Iron during infection
| 2.4: | Avoid administering IV iron to patients with active systemic infections. (Not Graded) |
Chapter 3: Use of ESAs and other agents to treat anemia in CKD
ESA INITIATION
ESA MAINTENANCE THERAPY
ESA DOSING
ESA ADMINISTRATION
Frequency of administration
| 3.10: | We suggest determining the frequency of ESA administration based on CKD stage, treatment setting, efficacy considerations, patient tolerance and preference, and type of ESA. (2C) |
TYPE OF ESA
EVALUATING AND CORRECTING PERSISTENT FAILURE TO REACH OR MAINTAIN INTENDED HEMOGLOBIN CONCENTRATION
Frequency of monitoring
Initial ESA hyporesponsiveness
Subsequent ESA hyporesponsiveness
Management of poor ESA responsiveness
ADJUVANT THERAPIES
EVALUATION FOR PURE RED CELL APLASIA (PRCA)
Chapter 4: Red cell transfusion to treat anemia in CKD
USE OF RED CELL TRANSFUSION IN CHRONIC ANEMIA
URGENT TREATMENT OF ANEMIA
Chapter 1: Diagnosis and evaluation of anemia
TESTING FOR ANEMIA
BACKGROUND
In any individual, anemia may be the initial laboratory sign of an underlying medical problem. Consequently, a complete blood count, including the hemoglobin (Hb) concentration, is routinely part of global health assessment in most adults, whether or not they have chronic kidney disease (CKD). In patients with CKD but stable kidney function, the appearance or progression of anemia may herald a new problem that is causing blood loss or is interfering with red cell production. The anemia should be evaluated independently of CKD stage in order to identify any reversible process contributing to the anemia. The causes of acquired anemia are myriad and too many to include in a guideline such as this. A comprehensive list of causes and the approach to diagnosis can be found in a standard textbook of medicine or hematology. The most commonly encountered reversible cause of chronic anemia or worsening anemia in CKD patients, other than anemia related directly to CKD, is iron deficiency anemia.
Frequency of testing for anemia
RATIONALE
Relatively little is known about the development and progression of anemia in patients with CKD. Consequently, one cannot determine precisely the optimal frequency at which Hb levels should be monitored. The recommendation that patients with CKD be periodically evaluated for anemia rests on observations that, in the absence of use of erythropoiesis-stimulating agents (ESAs), there often is a gradual decline in Hb over time in patients with CKD as the level of glomerular filtration rate (GFR) declines,1 suggesting the need for regular surveillance of Hb concentration. The frequency of Hb monitoring, regardless of CKD stage, should be influenced by the Hb level (i.e., more frequent monitoring may be appropriate in patients with more severe anemia) and rate of decline in Hb level. As kidney function declines and in patients with more advanced CKD stages, the incidence and prevalence of anemia increases. Thus, in order to identify CKD patients who may need intervention with iron administration, an ESA, or even require a transfusion, more frequent monitoring of the Hb concentration will be necessary at later CKD stages.
More frequent monitoring is recommended for adult CKD 5HD and CKD 5PD patients with anemia who are not receiving an ESA; at least monthly in CKD 5HD patients and at least every 3 months in CKD 5PD patients. In CKD 5HD patients, Hb monitoring is traditionally performed prior to a mid-week hemodialysis (HD) session. While this is not essential it probably does tend to minimize Hb variability due to the longer inter-dialytic interval between the last treatment of one week and the first of the next. As in all patients, Hb testing should be performed whenever clinically indicated, such as after a major surgical procedure, hospitalization, or bleeding episode.
In the pediatric population with CKD, there is no direct evidence to recommend a different frequency of monitoring for anemia than for adults. In the Chronic Kidney Disease in Children Prospective Cohort Study (CKiD), which evaluated 340 North American children with CKD using iohexol-determined GFR,2 below a GFR threshold of 43 ml/min per 1.73m2, there was a linear relationship between Hb and GFR, with Hb 0.3 g/dl (3 g/l) lower per 5 ml/min per 1.73m2 lower GFR. Above that threshold, there was a nonsignificant association of 0.1 g/dl (1 g/l) lower Hb for every 5 ml/min per 1.73m2 lower GFR. Because serum creatinine-based estimated glomerular filtration rate (eGFR) using the Schwartz formula may overestimate the true GFR in the children3 providers need to consider the potential for Hb decline and anemia even at early stages of CKD and monitor accordingly. In children with CKD 5HD and CKD 5PD, monthly monitoring for anemia is standard clinical practice.
Diagnosis of anemia
RATIONALE
The Hb concentration values that define anemia and should lead to initiation of an evaluation for the cause of anemia are dependent on sex and age. The recommended Hb values for adults and children represent the World Health Organization (WHO) definition of anemia and establish a benchmark for anemia workup that has been applied across populations.4
An alternative source for Hb concentration values that define anemia in children between 1 and 19 years is based on US National Health and Nutrition Examination Survey III (NHANES III) data from 1988-945 (Table 1). For children between birth and 24 months, the data are taken from normal reference values6 (Table 2).
These thresholds for diagnosis of anemia and evaluation for the causes of anemia should not be interpreted as being thresholds for treatment of anemia. Rather than relying on a single laboratory test value, in patients without an apparent cause for a low Hb level, the value should be confirmed to be below the threshold values for diagnosis of anemia prior to initiating a diagnostic work up.
Table 1: Hb levels in children between 1-19 years for initiation of anemia workupa
| All races/ethnic groups | Number of subjects | Mean Hb g/dl (g/l) | Standard deviation g/dl (g/l) | Anemia definition met if value is <5th percentile g/dl (g/l) |
|---|---|---|---|---|
| Boys | ||||
| 1 yr and over | 12,623 | 14.7 (147) | 1.4 (14) | 12.1 (121) |
| 1-2 yr | 931 | 12.0 (120) | 0.8 (8) | 10.7 (107) |
| 3-5 yr | 1,281 | 12.4 (124) | 0.8 (8) | 11.2 (112) |
| 6-8 yr | 709 | 12.9 (129) | 0.8 (8) | 11.5 (115) |
| 9-11 yr | 773 | 13.3 (133) | 0.8 (8) | 12.0 (120) |
| 12-14 yr | 540 | 14.1 (141) | 1.1 (11) | 12.4 (124) |
| 15-19 yr | 836 | 15.1 (151) | 1.0 (10) | 13.5 (135) |
| Girls | ||||
| 1 yr and over | 13,749 | 13.2 (132) | 1.1 (11) | 11.4 (114) |
| 1-2 yr | 858 | 12.0 (120) | 0.8 (8) | 10.8 (108) |
| 3-5 yr | 1,337 | 12.4 (124) | 0.8 (8) | 11.1 (111) |
| 6-8 yr | 675 | 12.8 (128) | 0.8 (8) | 11.5 (115) |
| 9-11 yr | 734 | 13.1 (131) | 0.8 (8) | 11.9 (119) |
| 12-14 yrb | 621 | 13.3 (133) | 1.0 (10) | 11.7 (117) |
| 15-19 yrb | 950 | 13.2 (132) | 1.0 (10) | 11.5 (115) |
Hb, hemoglobin; yr, year.
aBased on NHANES III data, United States, 1988-94.5
bMenstrual losses contribute to lower mean and 5th percentile Hb values for group.
Table 2 | Hb levels in children between birth and 24 months for initiation of anemia workupa
| Age | Mean Hb (g/dl (g/l) | -2 SDb g/dl (g/l) |
|---|---|---|
| Term (cord blood) | 16.5 (165) | 13.5 (135) |
| 1-3 d | 18.5 (185) | 14.5 (145) |
| 1 wk | 17.5 (175) | 13.5 (135) |
| 2 wk | 16.5 (165) | 12.5 (125) |
| 1 mo | 14.0 (140) | 10.0 (100) |
| 2 mo | 11.5 (115) | 9.0 (90) |
| 3-6 mo | 11.5 (115) | 9.5 (95) |
| 6-24 mo | 12.0 (120) | 10.5 (15) |
d, day; Hb, hemoglobin; mo, month; SD, standard deviation; wk, week.
aData taken from normal reference values. This was published in Nathan DG, Orkin SH. Appendix 11: Normal hematologic values in children. In: Nathan DG, Orkin SH, Ginsburg D et al. (eds). Nathan and Oski's Hematology of Infancy and Childhood, 6th edn. p 1841, © Elsevier, 2003.6
bValues 2 standard deviations below the mean are equivalent to <2.5th percentile.
Investigation of anemia
RATIONALE
Complete blood count
The complete blood count (CBC) provides information about the severity of anemia and adequacy of bone marrow function. Severity of anemia is assessed best by measuring Hb concentration rather than hematocrit. The latter measurement is a relatively unstable analyte and its measurement lacks standardization and is instrumentation dependent, since it is derived indirectly by automated analyzers.7, 8, 9 There is no evidence to support any different recommendation for the initial evaluation of anemia for children compared to adults.
In addition to Hb concentration, other reported results of the CBC may convey important clinical information. The anemia of CKD is hypoproliferative, and in general, normochromic and normocytic. In this regard it is morphologically indistinguishable from the anemia of chronic disease.10 Folate or vitamin B12 deficiencies may lead to macrocytosis, whereas iron deficiency or inherited disorders of Hb formation (e.g., α- or β-thalassemia) may produce microcytosis. Iron deficiency, especially if longstanding, is associated with hypochromia (low mean corpuscular hemoglobin [MCH]). Macrocytosis with leucopenia or thrombocytopenia suggests a generalized disorder of hematopoiesis caused by toxins (e.g., alcohol), nutritional deficit (vitamin B12 or folate deficiency), or myelodysplasia. When these findings are present, further diagnostic evaluation may be indicated.
The low erythropoietic activity that characterizes the anemia of CKD is consistent with insufficient erythropoietin stimulation. Erythropoietin levels are not routinely used in distinguishing erythropoietin deficiency from other causes of anemia in patients with CKD in most clinical settings and their measurement is generally not recommended.11, 12 Effective erythropoietic proliferative activity is most simply assessed by determination of the absolute reticulocyte count. Abnormalities of the white blood cell count and differential or platelet count are not typical of the anemia of CKD and should prompt investigation for other processes.
Reticulocyte count, which may be obtained with automated CBC testing, may be high in patients who have active blood loss or hemolysis, and may be low in hypoproliferative erythropoiesis with anemia.
Iron Status
There are two important and distinct aspects of the assessment of iron status testing: the presence or absence of storage iron and the availability of iron to support ongoing erythropoiesis. The serum ferritin is the most commonly used test for evaluation of storage iron, for which the 'gold standard' remains examination of a bone marrow aspiration stained for iron.13 The transferrin saturation (TSAT; serum iron x 100 divided by total iron binding capacity) is the most commonly used measure of the availability of iron to support erythropoiesis. The serum ferritin is affected by inflammation and is an 'acute phase reactant'13 and, thus, ferritin values have to be interpreted with caution in CKD patients, especially those on dialysis in whom subclinical inflammation may be present.14
Serum ferritin values ≤30 ng/ml (≤30 µg/l) indicate severe iron deficiency and are highly predictive of absent iron stores in bone marrow.15, 16 Ferritin values >30 ng/ml (>30 µg/l), however, do not necessarily indicate the presence of normal or adequate bone marrow iron stores. Studies assessing ferritin levels above which all or nearly all patients with CKD have normal bone marrow iron stores have produced varied results but most CKD patients, including those who are on HD, will have normal bone marrow iron stores when their serum ferritin level is ≥300 ng/ml (≥300 µg/l). Even at serum ferritin levels of 100 ng/ml (100 µg/l) most CKD patients have stainable bone marrow iron stores.16, 17, 18, 19, 20, 21 As will be discussed in Chapter 2, the serum ferritin and TSAT values are often used together to assess iron status, diagnose iron deficiency, and predict an erythropoietic response to iron supplementation (Supplementary Table 1 online).
Other tests of iron status, such as percentage of hypochromic red blood cells and reticulocyte Hb content may be used instead of, or in addition to, TSAT and ferritin levels if available. Measurement of hepcidin levels has not been shown to be clinically useful or superior to more standard iron status tests in patients with CKD.22, 23
Vitamin B12 and folate
Folate and vitamin B12 deficiency are uncommon but important causes of treatable anemia, typically associated with macrocytic red blood cell (RBC) indices. Limited data indicate a prevalence of vitamin B12 and folate deficiency in ≤10% of HD patients; the prevalence in CKD patients is not known. Nonetheless, since these deficiencies are easily correctable, and in the case of vitamin B12 may indicate other underlying disease processes, assessment of folate and vitamin B12 levels are generally considered standard components of anemia evaluation, especially in the presence of macrocytosis. Folate deficiency is best detected in most patients with serum folate level testing; RBC folate levels can be measured when serum folate levels are equivocal or when there is concern that recent dietary intake may obscure underlying folate deficiency using serum levels alone.24
Additional tests
Other tests, in addition to those indicated above, may be appropriate in individual patients and in certain specific clinical settings. For instance measurement of high sensitivity C-reactive protein (CRP) may be indicated if occult inflammation is a concern. In certain countries and/or in patients of specific nationalities or ethnicities, testing for hemoglobinopathies, parasites, and other conditions may be appropriate.
DISCLAIMER
While every effort is made by the publishers, editorial board, and ISN to see that no inaccurate or misleading data, opinion or statement appears in this Journal, they wish to make it clear that the data and opinions appearing in the articles and advertisements herein are the responsibility of the contributor, copyright holder, or advertiser concerned. Accordingly, the publishers and the ISN, the editorial board and their respective employers, office and agents accept no liability whatsoever for the consequences of any such inaccurate or misleading data, opinion or statement. While every effort is made to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this Journal, should only be followed in conjunction with the drug manufacturer's own published literature.
SUPPLEMENTARY MATERIAL
Supplemental Table 1: Association between iron status and level of
anemia in multivariable analyses.
Supplementary material is linked to the online version of the paper at
http://www.kdigo.org/clinical_practice_guidelines/anemia.php
Chapter 2: Use of iron to treat anemia in CKD
TREATMENT WITH IRON AGENTS
BACKGROUND
Correction of iron deficiency with oral or intravenous iron supplementation can reduce the severity of anemia in patients with CKD.25, 26 Untreated iron deficiency is an important cause of hyporesponsiveness to ESA treatment.27, 28 It is important to diagnose iron deficiency because treatment can readily correct the associated anemia and investigation for the cause of iron deficiency, which should follow its detection, can lead to important diagnoses. In the absence of menstrual bleeding, iron depletion and iron deficiency usually result from blood loss from the gastrointestinal tract. There are additional considerations in CKD patients with iron deficiency. For instance, hemodialysis patients are subject to repeated blood loss due to retention of blood in the dialyzer and blood lines. Other contributing causes in hemodialysis and other CKD patients include frequent blood sampling for laboratory testing, blood loss from surgical procedures (such as creation of vascular access), interference with iron absorption due to medications such as gastric acid inhibitors and phosphate binders, and reduced iron absorption due to inflammation.29 The reader is referred to standard textbooks of medicine and pediatrics for more extensive discussions on the diagnosis and evaluation of patients with known or suspected iron deficiency.
Iron supplementation is widely used in CKD patients to treat iron deficiency, prevent its development in ESA-treated patients, raise Hb levels in the presence or absence of ESA treatment, and reduce ESA doses in patients receiving ESA treatment. Iron administration is appropriate when bone marrow iron stores are depleted or in patients who are likely to have a clinically meaningful erythropoietic response. It is prudent, however to avoid iron therapy in patients in whom it is unlikely to provide meaningful clinical benefit, i.e., avoid transfusion and reduce anemia-related symptoms, and in those in whom potential benefit is outweighed by risks of treatment.23, 30, 31, 32 There are relatively few data on the long-term clinical benefits of iron supplementation other than direct effects on the Hb concentration. There is similarly little information on the long-term adverse consequences of iron supplementation in excess of that necessary to provide adequate bone marrow iron stores.33, 34, 35 Since bone marrow aspiration for assessment of iron stores is rarely done in clinical practice, iron supplementation is typically assessed by blood-based iron status tests without knowledge of bone marrow iron stores.27, 28, 36, 37, 38
The following statements provide recommendations for use of iron supplementation in patients with CKD.
*Based on patient symptoms and overall clinical goals, including avoidance of transfusion, improvement in anemia-related symptoms, and after exclusion of active infection.
**Consistent with Recommendations #3.4.2 and 3.4.3.
***Based on patient symptoms and overall clinical goals including avoidance of transfusion and improvement in anemia-related symptoms, and after exclusion of active infection and other causes of ESA hyporesponsiveness.
RATIONALE
In patients with CKD-associated anemia, iron supplementation is intended to assure adequate iron stores for erythropoiesis, correct iron deficiency, and, in patients receiving ESA treatment, prevent iron deficiency from developing. Iron supplementation, particularly with intravenous iron, can enhance erythropoiesis and raise Hb levels in CKD patients with anemia even when TSAT and ferritin levels are not indicative of absolute iron deficiency, and even when bone marrow studies reveal adequate iron stores.38, 39, 40 Iron treatment, particularly when administered intravenously, has also been consistently demonstrated to improve the erythropoietic response to ESA treatment.27, 28, 32, 36, 37, 41, 42, 43 For any individual patient the optimal balance of Hb level, ESA dose, and iron dose at which clinical benefit is maximized and potential risk is minimized is not known. Prescribing iron therapy for CKD patients is complicated by the relatively poor diagnostic utility of serum ferritin and TSAT tests to estimate body iron stores or for predicting a Hb response to iron supplementation.23, 30 Even examination of bone marrow iron stores, considered the 'gold standard' for assessment of iron stores, does not predict erythropoietic responsiveness to iron supplementation in patients with CKD with a high degree of accuracy.16, 23, 30, 40 It is important that the short and long-term safety of oral and intravenous (IV) iron agents, when known, be carefully considered when iron therapy is prescribed, and that the potential for as yet undiscovered toxicities also be taken into account. In each patient there must be consideration of current and desired Hb level, ESA dose and trends in ESA dose over time, assessment of the Hb response to iron supplementation, ongoing blood loss, and changes in iron status tests. While observational studies have not for the most part produced strong evidence of significant toxicity of chronic IV iron administration, the clinical benefit of such treatment has also not been convincingly demonstrated, although a recent randomized controlled trial (RCT) in patients with heart failure (some of whom also had mild CKD) is encouraging.44
TSAT and ferritin levels
The two most widely available tests for assessing iron status are the TSAT and serum ferritin level. A very low serum ferritin (<30 ng/ml [<30 µg/l]) is indicative of iron deficiency.16 Except in this circumstance, the TSAT and serum ferritin level have only limited sensitivity and specificity in patients with CKD for prediction of bone marrow iron stores and erythropoietic response to iron supplementation 16, 17, 18, 19, 20, 21, 40, 45 (Figures 1 and 2). Their utility is further compromised by substantial inter-patient variability unrelated to changes in iron store status.46
Evidence to support a recommendation for specific TSAT and ferritin levels at which iron therapy should be initiated or as 'targets' for iron therapy is limited, with very few RCTs.16, 17, 18, 19, 20, 21 No iron intervention trials have been sufficiently powered or of long enough duration to assess long-term safety and no studies have addressed the clinical benefit, cost effectiveness, and risk-benefit comparison of using different TSAT and ferritin levels for the diagnosis of iron deficiency or as a trigger for iron supplementation.
The Work Group sought to recommend iron targets that balance diagnostic sensitivity and specificity with assumptions regarding safety. Previous clinical practice recommendations (Kidney Diseae Outcomes Quality Initiative [KDOQI] 2006 and others), largely opinion-based, indicated that supplemental iron should be administered to maintain ferritin levels >200 ng/ml (>200 µg/l) in CKD 5HD patients and >100 ng/ml (>100 µg/l) in CKD ND and CKD 5PD with TSAT >20% in all CKD patients. These guidelines also indicated that there was insufficient evidence to recommend routine IV iron administration when the ferritin level was >500 ng/ml (>500 µg/l).
Most CKD patients with serum ferritin levels >100 ng/ml (>100 µg/l) have normal bone marrow iron stores,16, 17, 18, 19, 20, 21 yet many such patients will also have an increase in Hb concentration and/or reduction in ESA dose if supplemental iron is provided.16, 23, 30, 31, 40, 45 A substantial fraction of CKD patients with anemia and TSAT >20% respond to iron supplementation with an increase in Hb concentration and/or reduction in ESA dose. Therefore, for patients who have not been receiving iron supplementation, we suggest iron administration in anemic CKD patients with TSAT <30% and serum ferritin <500 ng/ml (<500 µg/l) if an increase in Hb level is desired, particularly if intended to avoid transfusions and reduce anemia-related symptoms, and/or reduction in ESA dose, after consideration of the potential risks of iron administration. The safety of providing additional iron to intentionally maintain TSAT >30% and serum ferritin >500 ng/ml (>500 µg/l) has been studied in very few patients. We do not recommend routine use of iron supplementation in patients with TSAT >30% or serum ferritin >500 ng/ml (>500 µg/l) since, as stated above, the benefits and risks of doing so are inadequately studied. In all patients receiving iron, it is important to weigh both short-term and acute toxicities associated with iron therapy and exclude the presence of active infection (Recommendation 2.4) before embarking on a course of IV iron treatment.
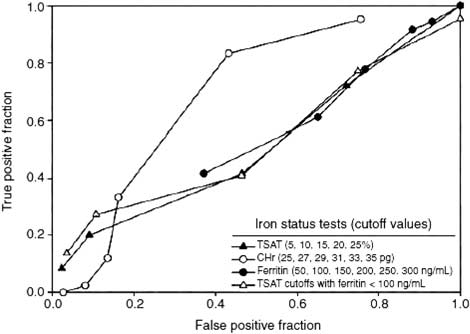
Figure 1 | Receiver operating characteristic (ROC) curves, examining the utility of iron status tests to distinguish iron deficient from non-deficient study patients. Reprinted with permission from Macmillan Publishers Ltd: Kidney International. Van Wyck DB, Roppolo M, Martinez CO et al. A randomized, controlled trial comparing IV iron sucrose to oral iron in anemic patients with nondialysis-dependent CKD. Kidney Int 2005; 68: 2846-2856; 45 accessed http://www.nature.com/ki/journal/v68/n6/full/4495631a.html.
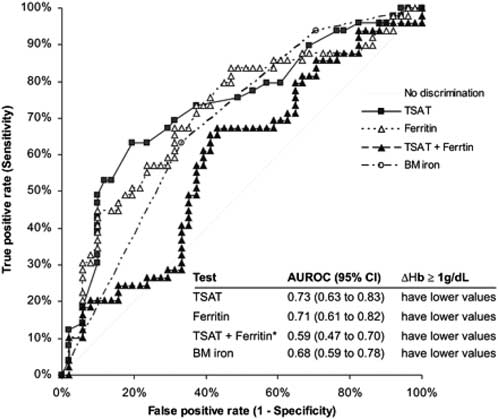
Figure 2 | Sensitivity and specificity of TSAT and serum ferritin (ferritin) and their combination (TSAT + ferritin) and bone marrow iron (BM iron) to identify correctly a positive erythropoietic response (≥1-g/dl [≥10-g/l] increase in Hb [ΔHb]) to intravenous iron in 100 nondialysis patients with CKD (areas under the ROCs). Reproduced with permission from American Society of Nephrology40 from Stancu S, Barsan L, Stanciu A et al. Can the response to iron therapy be predicted in anemic nondialysis patients with chronic kidney disease? Clin J Am Soc Nephrol 2010; 5: 409-416; permission conveyed through Copyright Clearance Center; accessed http: http://cjasn.asnjournals.org/content/5/3/409.long
There is only very limited evidence in patients with CKD that informs any decision about defining any specific upper limits for iron status targets in guiding iron treatment.47, 48 Previous guidelines, such as the 2006 KDOQI guidelines and others, have specified serum ferritin levels above which additional IV iron therapy was generally not recommended, 8, 49, 50, 51, 52 usually citing limits of 500-800 ng/ml (500-800 µg/l). However, no RCTs and few other studies have examined the efficacy and safety of providing IV iron to maintain ferritin levels >500-800 ng/ml (>500-800 µg/l). Most studies are retrospective and the few prospective studies have had small numbers of patients and short follow up, using surrogate outcomes such as Hb and ESA dose rather than more meaningful patient outcomes such as infection risk and mortality. In most patients with TSAT >30% or serum ferritin >500 ng/ml (>500 µg/l), any erythropoietic responsive to iron supplementation alone (i.e., the incremental change in Hb and/or reduction in ESA dose) will be small. In one RCT conducted in CKD 5HD patients with anemia, serum ferritin 500-1200 ng/ml (500-1200 µg/l), and TSAT<25%, patients received a 25% increase in epoetin dose and were randomly assigned to receive either no iron (control) or 1000 mg IV iron. At 6 weeks, Hb increased to a greater extent in the IV iron group.53 This study was not considered in the choice of target levels for ferritin and TSAT in this guideline in part because it studied only a restricted group of patients, all of whom also received an increase in ESA dose. The number of patients was too small and the period of observation too short to assess either clinically important outcomes or toxicity in a meaningful way (Supplementary Tables 2-4 online).
High ferritin levels in some studies have been associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute phase reactant is not clear. At increasingly higher ferritin levels, there is some evidence to indicate that hepatic deposition of iron increases.54, 55 Clinical sequelae of this have not been documented although such hepatic iron deposition might be of particular concern in patients with hepatitis C virus (HCV) infection.56 While some data are available linking ferritin levels in patients with hemochromatosis and transfusional tissue iron deposition in patients without CKD,57 it is not clear to what extent these findings are relevant to CKD patients or should be used to guide clinical practice in CKD patients.
Rather than focusing on serum ferritin levels as a predictor of outcomes, some observational studies have examined associations between patient outcomes and amount of iron administered. One such study found no adverse association between 2-year survival when the IV iron dose over 6 months was ≤1000mg, but a statistically significant higher mortality for iron doses >1000mg (adjusted hazards ratio [HR] 1.09; 95% confidence interval [CI] 1.01-1.17 for > 1000mg to 1800mg and 1.18; 95% CI 1.09-1.27 for > 1800mg).33 However, after using multivariable models accounting for time-varying measures of iron administration and other parameters, there was no statistically significant association between any level of iron administration and mortality. Another retrospective study using time-dependent and multivariate adjustment for case mix found that IV iron doses up to 400mg/month were associated with lower death rates compared to doses >400mg/month35 (Supplementary Table 5 online).
It is the consensus of the Work Group that additional IV iron should not routinely be administered in patients with serum ferritin levels that are consistently >500 ng/ml (>500 µg/l). In patients with Hb below the desired level who are receiving relatively high ESA doses, or in whom discontinuation of ESA therapy is preferred (for instance a CKD patient with malignancy), a therapeutic trial of additional IV iron (i.e., a single course of up to 1000mg over a period of several weeks which can be repeated as needed) may be undertaken in patients with serum ferritin levels >500 ng/ml (>500 µg/l) after due consideration of potential acute toxicities and long-term risks. Subsequent treatment decisions should be based on the patient's clinical status, including trends in TSAT, ferritin, and Hb level, and ESA dose and responsiveness.
Ferritin levels need to be interpreted with caution in patients who may have an underlying inflammatory condition as they may not predict iron stores or responsiveness to iron therapy in a manner similar to that when inflammation is absent. In the absence of a clinically evident infectious or inflammatory process, assessment of CRP may suggest the presence of an occult inflammatory state that may be associated with an elevated ferritin level and ESA-hyporesponsiveness (Supplementary Table 6 online).
Other iron status tests not as widely available as TSAT and ferritin such as percentage of hypochromic red cells, reticulocyte Hb content, zinc protoporphyrin, and soluble transferrin receptors may be used to assess iron status, but are less well studied.22, 23
There is no evidence that a higher ferritin target of 200 ng/ml (200 µg/l) is the appropriate or inappropriate cutoff in CKD 5HD pediatric patients. Consequently no change has been made to the 2006 KDOQI guideline in children with CKD and anemia, which recommended a ferritin target greater than 100 ng/ml (100 µg/l) for CKD 5HD, as well as for CKD 5PD and CKD ND who are not on ESA therapy.58
Iron Treatment
A decision to provide an individual patient with iron therapy should be based on an assessment that an increase in Hb level is desirable, that is, to avoid transfusions or reduce anemiarelated symptoms, and that the potential adverse effects of iron supplementation, either oral or IV, have been considered and are appropriately outweighed by the expected treatment benefit. Such supplementation could be in the form of oral or intravenous iron. Use of intramuscular iron has largely been abandoned. Each route has its own potential advantages and disadvantages. Oral iron is inexpensive, readily available, and does not require IV access, a particular concern in CKD patients not on HD. It is also not associated with severe adverse effects but gastrointestinal side effects are common and may limit adherence. This, along with variable gastrointestinal tract absorption, limits the efficacy of oral iron. IV iron avoids concerns about medication adherence and efficacy in treating iron deficiency, but requires IV access and has been associated with infrequent but severe adverse reactions. Decisions about the preferred route of iron supplementation should take into consideration severity of anemia and iron deficiency, the response, tolerance and adherence to prior oral iron administration, costs, and ease of obtaining venous access balanced against the desire to preserve venous access sites.
In patients with CKD ND, the available evidence supports an efficacy advantage of IV compared with oral administration of iron although the effect is rather small, with a weighted mean Hb difference of 0.31 g/dl (3.1 g/l).45, 59, 60, 61, 62, 63 Whether the small Hb benefit of IV iron in CKD ND patients is clinically meaningful or justifies the small risk of serious adverse events and unknown long-term risks is uncertain. The consensus of the Work Group is that a clearly defined advantage or preference for IV compared to oral iron was not supported by available evidence in CKD ND patients. Therefore, in such patients, the route of iron administration can be either IV or oral. In some patients the desire to avoid venipuncture (and preserve IV access) may favor in some patients, particularly those with mild iron deficiency, an initial trial of oral iron.
Oral iron is typically prescribed to provide approximately 200 mg of elemental iron daily (for instance ferrous sulfate 325 mg three times daily; each pill provides 65mg elemental iron). Smaller daily doses may be useful and better tolerated in some patients. Although ferrous sulfate is commonly available and inexpensive, other oral iron preparations may also be used; there is not significant evidence to suggest that other oral iron formulations are more effective or associated with fewer adverse side effects than ferrous sulfate. If the goals of iron supplementation are not met with a 1-3 month course of oral iron, it is appropriate to consider IV iron supplementation in a manner consistent with the above recommendation statements and the discussion that follows.
There is evidence supporting a preference for the IV route of iron administration in CKD 5HD patients derived from RCTs and other studies comparing IV iron with oral iron and placebo, with and without concomitant ESA treatment.27, 32, 62, 64, 65 In most of these studies, IV iron administration led to a greater increase in Hb concentration, a lower ESA dose, or both. In CKD 5HD patients, the ready IV access and convenience of being able to administer IV iron during HD treatments further supports the preference for the IV route for iron administration in these patients.
In prior CKD anemia guidelines,50 CKD 5PD patients were considered more similar to CKD ND than CKD 5HD in their need for and likely responsiveness to iron, as well as in their absence of ready venous access for IV iron administration. Limited studies of iron administration in CKD 5PD patients indicate that oral iron is of limited efficacy and that IV iron is superior to oral iron in terms of achieved Hb level and ESA dose. Consequently, this route is preferred in these patients, although the desire to preserve potential future venous access sites must be considered in such patients.66, 67, 68, 69, 70
IV iron may be provided as a single large dose or as repeated smaller doses depending on the specific IV iron preparation used (with the highest single dose varying by specific formulation). It is common practice to provide an initial course of IV iron amounting to approximately 1000 mg; this may be repeated if an initial dose fails to increase Hb level and/or allow a decrease in ESA dose and if the TSAT remains ≤30% and serum ferritin remains ≤500 ng/ml (≤500 µg/l).38
Decisions regarding continued iron therapy should take into consideration recent patient responses to iron therapy, iron status tests (TSAT and ferritin), Hb concentration, ESA responsiveness and ESA dose in ESA-treated patients, ongoing blood losses, trends in each parameter, and the patient's clinical status. Serum ferritin and TSAT levels should not be measured until at least one week has elapsed since the most recent prior IV iron dose. Consideration of expected iron needs and evaluation for ongoing iron losses should precede further IV iron administration. Blood loss should be minimal in CKD ND and CKD 5PD patients, while CKD 5HD patients have reported to lose between 1-2 gm of iron per year related to the HD procedure and related circumstances.71, 72, 73 Thus, an apparent ongoing need for any iron supplementation in CKD ND and CKD 5PD patients or for more than 1-2 gm/yr in CKD 5HD patients should prompt assessment for a source of active blood loss. The need to consider trends in iron status tests are highlighted by consideration of a patient with decreasing TSAT and ferritin levels which may signify the presence of gastrointestinal bleeding or excessive dialysis-associated blood loss. As another example, an increasing TSAT and ferritin level may indicate excessive iron supplementation and a need to decrease or discontinue iron administration. Finally, an increase in ferritin level accompanied by a decrease in TSAT and Hb level suggests inflammation-mediated reticuloendothelial blockade.14
There are two commonly used approaches to ongoing or maintenance IV iron treatment in CKD 5HD patients: (1) periodic iron repletion, consisting of a series of IV iron doses administered episodically to replenish iron stores whenever iron status tests indicate the likelihood of iron deficiency or decrease below specific target levels; or (2) maintenance treatment, consisting of smaller doses administered at regular intervals to maintain iron status tests stable within specific limits with the intent of avoiding iron deficiency or decline of iron test parameters below specific levels. Limited evidence suggests that regular maintenance IV iron administration in CKD 5HD is associated with use of lower ESA doses and may result in lower cumulative iron doses41, 74, 75 but these data are insufficient to support a recommendation favoring any particular IV iron dosing strategy in this patient population. By nature of the clinical encounters with CKD 5PD patients, IV iron supplementation is often provided at periodic (e.g., monthly) visits.
Overall, the TSAT and ferritin recommendations above are applicable to children with CKD on ESA therapy. However, there is no evidence that a higher ferritin target of 200 ng/ml (200 µg/l) is the appropriate or inappropriate cutoff in pediatric CKD HD patients. Consequently no change has been made to the 2006 KDOQI guideline in CKD in children with anemia, which recommended a ferritin target greater than 100 ng/ml (100 µg/l) for CKD 5HD, as well as for CKD 5PD and CKD ND who are on ESA therapy.58
IRON STATUS EVALUATION
RATIONALE
In the absence of clinical trials that specifically inform the optimal frequency for testing of iron status, and consistent with prior guidelines,50 the consensus of the Work Group is that patients who are on ESA therapy, regardless of whether iron treatment is also being used, should have tests of iron status at least every 3 months. Falling TSAT and/or ferritin levels are likely to reflect ongoing blood loss or consumption of available iron stores, and can be used to anticipate the need for future or additional iron supplementation. In patients on oral iron treatment, iron status testing can also be used to assess adherence with iron treatment. Increasing TSAT and/or ferritin levels may indicate that iron treatment is excessive and can be stopped or reduced. Increasing ferritin levels in association with stable or declining TSAT levels may also indicate the presence of inflammation, infection, or other clinical situations inducing acute phase reactants during which time the appropriateness of continued iron administration may need to be reassessed.14
In some circumstances, more frequent iron status testing may be appropriate, including following initiation of ESA or iron therapy or when the ESA dose or dose frequency is increased. Iron status testing is also important in the assessment of patients who become less responsive to ESA treatment.
Despite the absence of specific data in the pediatric CKD population, this recommendation is considered applicable to children since there are no reasons to suggest a different recommendation. Since the 2006 KDOQI guideline for anemia in pediatric CKD,58 no new evidence regarding iron therapy for children with CKD has been published. The suggestion for oral iron supplementation in children is 2-6 mg/kg/day of elemental iron in 2-3 divided doses.76, 77 An RCT of 35 iron replete pediatric CKD 5HD patients evaluated their response to either weekly IV iron dextran dosed by weight or oral iron 6 mg/kg/day. Only the IV iron dextran produced a significant increase in the serum ferritin levels and showed a significant decrease in ESA dose required to maintain target Hb levels.78 An international multicenter double-blind RCT investigated the safety and efficacy of two dosing regimens (1.5 mg/kg or 3 mg/kg) of ferric gluconate in iron-deficient pediatric hemodialysis patients receiving concomitant ESA therapy. Efficacy and safety profiles were comparable, with no unexpected adverse events with either dose.79 Based on this trial, the recommendation for initial ferric gluconate therapy is 1.5 mg/kg for eight doses for iron-deficient pediatric CKD 5HD patients and 1 mg/kg per week for iron-replete pediatric CKD 5HD patients, with subsequent dose adjustments made according to TSAT and/or ferritin levels.79, 80 Iron sucrose has also been used in children with CKD81 but, as of yet, no RCTs have been published in this population. Although it is not uncommon that pediatric CKD 5PD and CKD ND patients either do not respond to or tolerate oral iron therapy, the need for IV access for parenteral iron therapy often limits its utilization in children.
CAUTIONS REGARDING IRON THERAPY
RATIONALE
Any form of IV iron may be associated with potentially severe acute reactions.82, 83, 84, 85, 86, 87, 88, 89, 90, 91 The symptoms of most concern are hypotension and dyspnea, which in the worst cases may be catastrophic with features of anaphylaxis. The cause of reactions has not been fully characterized, but may involve immune mechanisms and/or release of free, reactive iron into the circulation with induction of oxidative stress. The mechanisms of acute reactions may differ for different iron preparations. Certain iron dextrans in particular have been associated with reactions characteristic of anaphylaxis. The rate of such reactions is estimated to occur in 0.6-0.7% of patients treated. The serious adverse effect event rate may be lower with low molecular weight iron dextran compared to high molecular weight iron dextran. 92, 93, 94, 95, 96
With non-dextran IV iron drugs, it is believed that anaphylactoid and other severe and potentially life-threatening reactions are less common, but this has not been well substantiated. Serious reactions including profound hypotension do occur, even if uncommonly, with all non-dextran IV iron preparations. Because all forms of IV iron drugs can be associated with serious immediate reactions, they should be used with vigilance. Since the rate of such reactions may be greater for iron dextran drugs we recommend that resuscitative medications and personnel trained to evaluate and treat serious adverse reactions be available when the initial dose of IV iron dextran is administered. The data to support such a recommendation for the initial dose of non-iron dextran compounds is not as strong. In the US, the Food and Drug Administration (FDA)-mandated labeling for ferumoxytol specifies that patients be observed for 60 minutes after administration. This may be reasonable advice for all IV iron drugs, including other new iron preparations such ferric carboxymaltose and iron isomaltoside. For each IV iron preparation prescribing physicians should be familiar with the drug's safety and toxicity profile and the product labeling warnings and recommendations for administration, as well as patient monitoring during and after treatment.
Iron during infection
| 2.4: | Avoid administering IV iron to patients with active systemic infections. (Not Graded) |
RATIONALE
Iron is essential for the growth and proliferation of most pathogens including many bacteria, viruses, fungi, parasites and helminthes, and also exerts subtle effects on immune function and host responses towards microbes.97 There is theoretical and experimental evidence to suggest that iron administration may worsen an existing infection but clinical evidence is lacking. In animal models, iron overload results in an impaired control of infections, specifically with intracellular bacteria or fungi.98, 99, 100, 101 In humans, tissue iron overload has been considered as a risk factor for the acquisition of certain infections and for an unfavorable clinical course of the infection. Data in CKD patients are conflicting.102, 103, 104 Since current evidence cannot provide a clear answer as to whether specific CKD patient groups are at increased risk for infection, or of having a poorer outcome with infection when anemia is treated with IV iron, the Work Group suggests that IV iron not be administered when patients have an active systemic infection. Clinical judgment is necessary in each individual patient to assess whether there is an immediate need for IV iron (as opposed to delaying treatment until resolution of an infection), likelihood of achieving benefit from a dose of IV iron in the setting of an active infection, and the severity of an infection.
RESEARCH RECOMMENDATIONS
Much regarding the testing of iron status and use of iron supplementation, particularly IV, in CKD patients of all stages remains unknown. There is a serious lack of large, prospective clinical trials with assessment of clinically meaningful outcomes and toxicities; rather, most have been small,short-term studies focusing primarily on surrogate outcomes such as increase in Hb level and reduction in ESA dose. Some important questions that should be addressed in future studies might include:
- What is the comparative risk-benefit balance of various treatment strategies that include differing ratios of ESA dosing and iron supplementation to achieve a particular Hb level?
- Is there a role, and if so under what circumstances, for anemia management in CKD patients with iron alone, without ESA treatment (or with only ESA 'rescue therapy' for particularly low Hb levels)?
- Is there important long-term toxicity of IV iron supplementation and if so, under what circumstances and in what CKD patient groups?
- Is IV iron administration, with or without concomitant ESA dose increases, safe and of clinical benefit, in patients with ferritin levels >500-800 ng/ml (>500-800µg/l)?
- What are the best laboratory tests to guide decisions regarding initiation, ongoing treatment, and discontinuation of iron supplementation?
- Is current iron and anemia management in pediatric CKD patients appropriate?
DISCLAIMER
While every effort is made by the publishers, editorial board, and ISN to see that no inaccurate or misleading data, opinion or statement appears in this Journal, they wish to make it clear that the data and opinions appearing in the articles and advertisements herein are the responsibility of the contributor, copyright holder, or advertiser concerned. Accordingly, the publishers and the ISN, the editorial board and their respective employers, office and agents accept no liability whatsoever for the consequences of any such inaccurate or misleading data, opinion or statement. While every effort is made to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this Journal, should only be followed in conjunction with the drug manufacturer’s own published literature.
SUPPLEMENTARY MATERIAL
Supplemental Table 2: Summary table of RCT examining the effect of IV iron ? EPO vs. EPO only in patients with HD-CKD (categorical outcomes).Supplemental Table 3: Summary table of RCT examining the effect of IV iron ? EPO vs. EPO only in patients with HD-CKD (continuous outcomes).
Supplemental Table 4: Summary table of adverse events in RCT examining the effect of IV iron ? EPO vs. EPO only in patients with HD-CKD (continuous outcomes).
Supplemental Table 5: Association between cumulative iron dose and clinical outcome in multivariable analyses.
Supplemental Table 6: Association between iron status and clinical outcome in multivariable analyses.
Supplementary material is linked to the online version of the paper at http://www.kdigo.org/clinical_practice_guidelines/anemia.php
Chapter 3: Use of ESAs and other agents* to treat anemia in CKD
ESA INITIATION
BACKGROUND
The introduction of recombinant human erythropoietin (rHuEPO) into clinical practice in the 1980 s was a major breakthrough in the treatment of the anemia of patients with CKD. The development of rHuEPO was aimed at replacing the insufficient endogenous erythropoietin (EPO) production related to CKD progression. It remains unclear whether the main cause of anemia is a loss of kidney EPO production capacity or a derangement in oxygen sensing, as proposed more recently.105
In the early years, rHuEPO administration was regarded by the nephrology community as a beneficial therapy for long-term dialysis patients whose Hb values fell to extremely low levels, making them transfusion-dependent. The immediate benefit of rHuEPO in CKD patients with severe anemia and anemia-related signs and symptoms was clear. In addition, the reduction in the need for regular blood transfusions was another major benefit, resulting in less frequent transmission of blood-borne viral diseases, such as hepatitis B and C, less allosensitization, predisposing to prolonged wait times or failure to receive a kidney transplant, transplant rejection, and less transfusional hemosiderosis.106, 107, 108, 109
After introduction of rHuEPO into clinical practice its administration was limited to dialysis patients with the most severe forms of anemia. Progressively, its use was extended to the majority of dialysis patients with renal anemia, and subsequently also to anemic patients with CKD 4-5 in countries in which the high cost of rHuEPO did not limit the number of patients eligible for this treatment.
Hb targets also increased progressively, often into the range of normal values. The idea that anemia should be corrected completely was based on pathophysiologic considerations and the demonstration by numerous observational studies of an inverse association between Hb concentrations up into the normal range and intermediate outcomes such as left ventricular hypertrophy,110 as well as hard patient outcomes such as cardiovascular events,111, 112, 113 hospital admission,114 and death.115, 116 Of note, a recent study also showed that CKD 5D patients with naturally occurring Hb concentrations greater than 12 g/dl (120 g/l) were not at increased mortality risk.117 However, the suggestion drawn from epidemiological studies that anemia should be completely corrected in patients with CKD was not supported by the Normal Hematocrit Study in CKD 5D patients118 and several recent randomized controlled trials (RCTs) performed in large CKD patient cohorts (Supplementary Table 7 online).
In CKD 5D patients Hb concentrations often fall below 8 g/dl (80 g/l) if anemia is untreated, whereas in CKD ND patients higher Hb concentrations are usual, unless patients are close to dialysis or have another contributing cause. The decision to prescribe ESAs should be based on evidence accrued from RCTs. However substantial heterogeneity exists in RCTs performed to evaluate ESA therapy, particularly in relation to classification of patients, research design, baseline Hb, target Hb, clinical outcome measures, and definitions of clinically meaningful improvements.
Outcomes of interest in RCTs of ESAs include mortality, cardiovascular and kidney endpoints, safety, quality of life (QoL), blood transfusions and cost. QoL outcomes are particularly important for CKD 5D patients and for some may be more important than cardiovascular events or mortality, since they have relatively short life expectancy and the symptoms attributable to anemia (e.g., low energy, fatigue, decreased physical function, and low exercise capacity) occur frequently and can be disabling.119 However, QoL is extremely difficult to quantify as is the clinical importance of changes measured. Furthermore, unless assessed under rigorous double-blind conditions, the validity of QoL measurements is questionable. Avoidance of transfusions is important, as mentioned above.
The guidelines to treat or not to treat the anemia of CKD are also valid for CKD 4-5T patients. Of note, blood transfusions may increase the risk of alloreactivity and rejection episodes after kidney transplantation.120 In addition a recent randomized trial has shown that early post-kidney transplant anemia correction by ESAs reduces the progression of allograft nephropathy, although its effect on hard outcomes in this patient population remains unknown.121
*Excluding iron which is discussed in Chapter 2.
| 3.1: | Address all correctable causes of anemia (including iron deficiency and inflammatory states) prior to initiation of ESA therapy. (Not Graded) |
RATIONALE
After diagnosing anemia in a patient with CKD all correctable causes should be treated before considering ESA therapy. Above all, this recommendation is based on the observation that iron supplementation given to CKD patients with proven iron deficiency or impaired iron availability ('functional iron deficiency') generally leads to an increase in Hb (See Chapter 2). However, the correction of other deficiency states also may ameliorate anemia. In patients with inflammatory diseases, including bacterial and viral infections, the attenuation of the inflammatory status is often followed by an improvement of Hb.
There are several reasons why correctable causes other than erythropoietin deficiency should be actively sought. As in any disease state, pathological conditions which can be cured should be corrected first. As examples, ESA treatment is unlikely to be fully effective in raising Hb concentrations until either severe systemic bacterial infections or severe secondary hyperparathyroidism are appropriately treated (Supplementary Table 8 online). When several different factors are thought to contribute to the anemia of CKD, even though the main underlying cause is impaired kidney EPO synthesis, appropriate medical care dictates treating all underlying causes.
RATIONALE
Treatment of severe anemia
Objective evidence to support treatment of Hb concentrations below 9 g/dl (90 g/l) is quite strong because the transfusion benefits are substantial and the QoL improvements are clinically important. However the safety of ESAs in treating severe anemia has not been evaluated in large placebo controlled trials.
The Canadian Erythropoietin Study Group reported a double-blind RCT of 118 CKD 5HD patients in 1990. ESA was utilized in patients with Hb concentrations <9 g/dl (<90 g/l), and three randomly allocated groups were followed (placebo, target Hb 9.5-11 g/dl [95-110 g/l], high target Hb >11 g/dl [>110 g/l]).122 Baseline Hb was 7.0 g/dl (70 g/l) and the mean transfusion requirement was 7 transfusions per year. After 8 weeks, 58% (N=23/40) in the placebo group were transfused and only 2.5% (N=1/40) was transfused in the group with target Hb of 9.5-11g/dl (95-110 g/l) and 2.6% (N=1/38) in the group with target Hb>11g/dl (>110 g/l). After 6 months, significant improvements in fatigue, physical function, and 6 minute walking tests were reported for the low Hb group compared to placebo, but no improvement was observed comparing low vs high Hb group. In an open-label RCT of only 83 CKD ND patients with Hb <10 g/dl (<100 g/l), significant improvements in energy and physical function were also reported.123
Treatment of moderate anemia
There are several large RCTs of ESA therapy where baseline Hb is >10 g/dl (>100 g/l). 118, 124, 125, 126, 127, 128 The intervention being tested in these trials is complete correction of anemia with ESAs, compared to partial correction with ESAs in five RCTs 118, 124, 125, 126, 128 and to placebo in one.127 A double-blind design is necessary to accurately assess subjective or clinician driven endpoints particularly QoL, starting dialysis, and giving transfusions. Notably, only 3 of the 6 trials were double-blind - the Normal Hematocrit Study reported in 1998,118 the Canada-Europe Study reported in 2005,126 and TREAT reported in 2009.127 The Scandinavian Study,125 CREATE124 and CHOIR128 trials were open label.
The US Normal Hematocrit Trial by Besarab et al.118 was the first of a series of RCTs which cast serious doubt on the assumption that full anemia correction should be achieved in the majority of dialysis patients. A cohort of 1233 prevalent CKD 5HD patients with symptomatic heart failure or ischemic heart disease were allocated to either partial treatment of anemia or full anemia correction, using epoetin-alfa. The eventually achieved hematocrit values were 31% and 40%, respectively. In the normal hematocrit group treated with epoetin there were 183 deaths and 19 myocardial infarcts, producing 202 primary events, compared to 164 events (150 deaths, 14 myocardial infarcts) in the group in which anemia was partially corrected with epoetin. The risk ratio for the primary endpoint was 1.3 (95% CI 0.9-1.9) which did not satisfy the pre-specified criterion for statistical significance (even though the nominal p value was 0.03) after adjusting for interim analyses. The trial was stopped early in a situation where the primary hypothesis was unlikely to be proven and the intervention being tested caused harm: 39% had vascular access clotting in the intervention arm and 29% in the control arm (P=0.001).
The double-blind Canada-Europe trial by Parfrey et al.126 of 596 incident CKD 5HD patients without symptomatic heart disease (18% with diabetic nephropathy) examined the question whether full anemia correction by epoetin-alfa in the group randomized to a Hb target of 13.5-14.5 g/dl (135-145 g/l), as compared to partial treatment of anemia in the group randomized to a Hb target of 9.5-11.5 g/dl (95-115 g/l), had a beneficial effect on left ventricular volume and mass index. The eventually achieved Hb values were 13.1 and 10.8 g/dl (131 and 108 g/l), respectively. There was no difference in left ventricular volume index or mass index between the two groups during this 96-week study. Of note, patients in the full anemia correction group had a significantly higher stroke incidence (secondary endpoint) than patients in the partial treatment correction group. However, the absolute numbers of patients with stroke were very small. As one might expect, the high Hb group received significantly fewer transfusions than the low Hb group, but extent of the benefit was modest: although 9% in the high Hb arm received at least one transfusion compared to 19% in the low Hb arm (P=0.004) during the 96-week study, the transfusions per patient per year was 0.3 in the high Hb arm and 0.7 in the low Hb arm (P<0.0001).129 In addition significant improvements in QoL were reported for the a priori selected domains of vitality and of fatigue.126, 130
The goal of the CREATE study by Drueke et al.124 was to show superiority of full anemia correction in terms of cardiovascular events, as compared to partial correction of anemia, when starting ESA therapy at an earlier stage than end-stage renal disease (ESRD). In this trial, 603 CKD 3-5 patients (26% with diabetes) were randomly allocated to either a Hb target of 13.0-15.0 g/dl (130-150 g/l) or a Hb target of 10.5-11.5 g/dl (105-115 g/l) using epoetin-beta. The eventually achieved Hb values were 13.5 and 11.6 g/dl (135 and 116 g/l), respectively. Dialysis was required in significantly more patients in the high Hb group than in the low Hb group. However the rate of fall of GFR in the two groups during the 3 year study was similar. Statistically significant improvements in some domains of QoL, including physical function and vitality, were observed in the high Hb group, although these must be interpreted cautiously because the study was open-label.
The US CHOIR study by Singh et al.128 similarly aimed to show superiority of full anemia correction by ESA administration in terms of cardiovascular events and death, as compared to partial treatment of anemia, in patients with CKD not yet on dialysis. In this trial, 1432 CKD 3-4 patients (49% with diabetes) were randomized to Hb targets of 13.5 g/dl (135 g/l) and 11.3 g/dl (113 g/l) using epoetin alfa. Withdrawal rate was high: 17% due to renal replacement therapy and 21% for other reasons. The study was prematurely stopped after an interim analysis with a median study duration of 16 months. The achieved Hb values were 12.6 and 11.3 g/dl (126 and 113 g/l), respectively. At this time point, 125 patients in the complete anemia correction group but only 97 patients in the standard correction group had reached the primary combined cardiovascular endpoint (P=0.03). No differences in QoL were observed comparing the two groups although, again, this finding must be interpreted cautiously because the study was open-label.
Finally, the international trial of darbepoetin-alfa in type 2 diabetes and CKD (TREAT) by Pfeffer et al.127 examined cardiovascular and kidney outcomes in 4038 CKD 3-4 patients. Of note, this is by far the largest ESA trial, and has the best research design, as it was placebo controlled and double-blinded. Patients received either darbepoetin-alfa to achieve a Hb target of 13.0 g/dl (130 g/l) or placebo with rescue darbepoetin-alfa when the Hb concentration was <9.0 g/dl (<90 g/l). The achieved Hb values were 12.5 and 10.6 g/dl (125 and 106 g/l), respectively. The median followup duration of the study was 29 months. There were no differences in the two primary endpoints, which were the composite outcomes of death or a cardiovascular event (first primary endpoint) and death or ESRD (second primary endpoint). The hazard ratio for death/composite cardiovascular event was 1.05 (95% CI 0.94-1.17), and for death or ESRD it was 1.06 (95% CI 0.96-1.19). However there was a substantial increased risk of stroke (HR 1.92; 95% CI 1.38-2.68), although the absolute risk of stroke overall was modest: 5.0% of the high Hb group had a stroke compared to 2.6% in the placebo group (P<0.001). The relative increase in risk of stroke was similar in patients with and without a past history of stroke. As a result, the absolute risk of stroke was substantial in the 11% of subjects with a prior history of stroke; 12% in the darbepoetin group compared to 4% in the placebo group. Venous thrombo-embolic events occurred significantly more frequently in the high Hb arm (2.0%) compared to the placebo arm (1.1%, P=0.02). A signal that normalization of Hb with darbepoetin may be harmful in patients with a history of malignancy was reported following a post-hoc analysis: 14/188 (7.4%) of those with a history of malignancy at baseline died from cancer in the darbepoetin arm compared to 1/160 (0.6%) (P=0.002) in the placebo arm. A statistically significant improvement in Functional Assessment of Cancer Therapy-Fatigue (FACT-fatigue) scores was reported at week 26 favoring the darbepoetin group, but the clinical significance of this was modest, as 55% of the high Hb group had a clinically important improvement in fatigue score compared to 50% of the placebo group. Transfusions were prescribed relatively frequently, and more often in the placebo arm (25%) compared to the high Hb arm (15%). The harm:benefit trade-off in TREAT was 1 stroke for 5 transfusions prevented by the high Hb target131 (Supplementary Tables 9-19 online). In a large subset of the TREAT patients QoL was assessed using FACT-fatigue, SF-36, and EQ-5D through 97 weeks. Compared to placebo, darbepoetin conferred a consistent, but small improvement over 97 weeks in fatigue and overall QoL, but none in energy and physical function. Interim stroke had a substantial negative impact on fatigue and physical function.132
Meta-analyses
Assessment of ESAs in CKD using meta-analysis is problematic because of the heterogeneity of patients entered, the different quality and research designs of the RCTs performed, and differences in definitions of endpoints. In addition abstraction of aggregate data from the reports of RCTs to populate the meta-analysis data base is also a limitation, as individual patient data would be preferable. The most recent meta-analysis133 concluded that higher Hb concentrations in CKD increases risk for stroke (relative risk [RR] 1.51, 95% CI 1.03-2.21), hypertension (RR 1.67, 95% CI 1.31-2.12), and vascular access thrombosis (RR 1.33; 95% CI 1.16-1.53), and may perhaps increase risk for death (RR 1.09; 95% CI 0.99-1.20), serious cardiovascular events (RR 1.15, 95% CI 0.98-1.33) or ESRD (RR 1.08; 95% CI 0.97-1.20). In our opinion, because of the heterogeneity of patients and interventions across studies in the meta-analysis greater credence should be given to the results of the very large, placebo controlled, double-blind trial, TREAT, than to the meta-analyses, in areas where the results differ: TREAT found no difference between the higher Hb, darbepoetin, group and the lower Hb, placebo, group for the two primary composite outcomes (either death or a cardiovascular event, or death or a renal event).127
The existing meta-analyses of QoL outcomes are further complicated by inclusion of data from open label studies, different instruments to measure QoL, differences in research design across RCTs, incomplete reporting as some trials chose (a priori) specific domains as trial outcomes, and differences in the definition of clinically meaningful improvement in QoL domains.119 Results from two systematic reviews published recently134, 135 suggest that improvements in QoL are maximized in the 10-12 g/dl (100-120 g/l) range. In CKD ND patients the review focused on energy and physical function134 and in CKD 5D patients the review focused on physical function and the meta-analysis on exercise tolerance.135
RATIONALE
The joint guideline from the American Society of Clinical Oncology136 and the American Society of Hematology137 recommend using ESA therapy with great caution in patients with active malignancy, particularly when cure is the anticipated outcome. This advice is supported in CKD patients by the post-hoc analysis in TREAT which demonstrated a significantly higher death rate from cancer in the darbepoetin arm in patients with a history of a malignant condition at baseline as compared with the placebo arm.127
The relative risk of stroke in patients in the darbepoetin arm of TREAT was the same in those with and without a history of stroke (i.e., approximately doubled). However the absolute risk of stroke was much higher in subjects with a history of stroke (in both study arms) and the absolute risk of stroke attributable to high Hb/darbepoetin was particularly high, 8% in those with a history of stroke vs 1% in those without a history of stroke over 29 months.138 Consequently the Work Group concluded that ESAs should be used with great caution in those with a prior history of stroke.
RATIONALE
In adult CKD-ND patients TREAT demonstrated that the high Hb darbepoetin arm was associated with harm. In the patients on placebo with rescue treatment allowed when Hb fell to below 9.0 g/dl (90 g/l) the achieved median Hb value was as high as 10.6 g/dl (106 g/l), despite the majority of patients receiving no or little darbepoetin127 (Supplementary Tables 15-19 online).
There is no convincing evidence that the active increase of Hb towards concentrations in the normal range leads to demonstrable benefit in adult patients with CKD stages 3-5. Moreover, when Hb falls below 10 g/dl (100 g/l) in these patients the Work Group were unconvinced that all patients should have an ESA initiated, particularly as the rate of Hb fall may be slow. It was suggested that the decision to initiate ESA therapy in CKD-ND when Hb is 49.0 and <10.0 g/dl (>90 and >100 g/l) should be individualized based on risk of requiring transfusions and on the presence of symptoms attributable to anemia, particularly as some patients may be at higher risk of requiring red-cell transfusions, and some patients are more prone to developing symptoms and signs associated with anemia (Supplementary Tables 15-19 online).
In adult hemodialysis patients the rate of fall of Hb is faster than in ND patients, and if untreated Hb will frequently fall below 8 g/dl (80 g/l).122 As the risk of transfusions is high in those HD patients whose Hb falls below 9 g/dl (90 g/l) the Work Group suggested that ESA therapy should be used to prevent the Hb concentration from falling below 9.0 g/dl (90 g/l), which in practice means that the Hb concentration at which ESA should be initiated should be between 9.0 and 10.0 g/dl [90 and 100 g/l] (Supplementary Tables 9-14 online).
However, there may be subgroups of adult CKD stage 3-5 and 5D patients in whom it may not be wise to let Hb values descend below 10 g/dl (100 g/l), particularly in elderly patients who are more prone to developing symptoms and signs associated with anemia, and those who are prone to requiring red-cell transfusions.
Moreover, physical and mental performances and QoL may be seriously compromised in adult CKD patients with severe anemia. RCTs supporting registration of epoetin-alfa for the treatment of anemia in dialysis patients demonstrated that ESA treatment of subjects with a Hb of <10 g/dl (<100 g/l) to a Hb target of approximately 10-12 g/dl (100-120 g/l) improved patient-reported physical functioning.134, 135 The question of the Hb value above which there is no further improvement in these parameters remains unsolved, especially for CKD-ND patients without diabetes and CKD-5D patients with or without diabetes.
In anemic children with CKD there are no RCTs examining the effects of ESA administration on hard outcomes. Therefore, any suggestion for Hb targets in this subgroup of CKD patients has to rely on results obtained in the adult CKD patient population and on clinical experience in the pediatric setting. The upper and lower Hb targets are opinion-based, in keeping with the lack of pediatric specific evidence. There are a number of factors unique to children that make exclusive reliance on evidence in adults inappropriate such as age-specific variation of normal Hb concentrations as well as QoL, growth, developmental, and psychological differences between children and adults.58 Limited data suggest that children with CKD and a Hb less than 9.9 g/dl (99 g/l) are at increased risk for mortality,139 left ventricular hypertrophy,140, 141 and/or decreased exercise capacity142 compared to those with a Hb greater than 9.9 g/dl (99 g/l). When evaluated as a continuous variable, hematocrit (Hct) was linked directly to measures of improved health and physical functioning in a health based QoL questionnaire administered to a pediatric CKD population.143
ESA MAINTENANCE THERAPY
RATIONALE
The suggestion to set the upper Hb target in general to values ≤11.5 g/dl (≤115 g/l) in adult CKD patients is based on the interpretation of the combined results of the recent major RCTs that there may be more harm than benefit at higher Hb concentrations. Of note, the update of the 2006 KDOQI anemia guideline in 2007 had already led to the recommendation to limit the upper Hb target to 12 g/dl (120 g/l), not to exceed 13 g/dl (130 g/l).51 The present suggestion not to exceed in general a Hb limit of 11.5 g/dl (115 g/l) has been influenced by the fact that the upper boundary of the Hb concentration in the control group of the major ESA RCTs usually did not exceed 11.5 g/dl (115 g/l); no data exist on the benefits of Hb targets between 11.5 and 13.0 g/dl (115 and 130 g/l); and high Hb targets are associated with adverse outcomes.
The Work Group recognized that some patients experience an improvement in QoL when the Hb value is above 11.5 g/dl (115 g/l). This opinion is supported by the heterogeneity of QoL outcomes in the major RCTs: in the double-blind Canada-Europe Study and in open label CREATE study statistically significant improvements in some QoL domains that may be clinically important were reported with higher Hb values.124, 126, 130 In the double-blind TREAT study the QoL benefits of higher Hb were modest127, 132 and in open label CHOIR study no benefits were observed128 (Supplementary Tables 9-19 online).
As all CKD patients in TREAT study also had type 2 diabetes, it is possible that improvements in QoL may be more difficult to achieve in this subgroup of patients than in those not suffering from diabetes.
An increase of Hb above 11.5 g/dl (115 g/l) towards 13 g/dl (130 g/l) may also be justified in individual patients with a high bleeding tendency since this results in lower transfusion needs, as shown by 8 RCTs.133
Obviously, increasing Hb above 11.5 g/dl (115 g/l) up to 13 g/dl (130 g/l) has to be weighed against the probability of increased harm. This perspective needs to be clearly explained to each patient who wishes to examine the possible benefits of more complete anemia correction.
| 3.6: | In all adult patients, we recommend that ESAs not be used to intentionally increase the Hb concentration above 13 g/dl (130 g/l). (1A) |
RATIONALE
The strong recommendation not to aim for Hb increases to concentrations >13 g/dl (>130 g/l) is based on the interpretation of the combined results of the recent major RCTs showing more harm than benefit with higher Hb targets, as compared to lower Hb targets, including increased risks for stroke,126, 127 hypertension,133 and vascular access thrombosis (in hemodialysis patients).118 TREAT did not demonstrate significant differences for serious cardiovascular or kidney events comparing correction of anemia with darbepoetin to the placebo group.127 Thus the increased risk of kidney events reported in CREATE124 and of cardiovascular events reported in CHOIR128 were not substantiated in the much larger TREAT trial.127 However, a recent meta-analysis point estimate indicated increased mortality at higher Hb target133 (Supplementary Tables 9-19 online).
An exception to the recommendation to avoid Hb increases to concentrations >13 g/dl (>130 g/l) might however be made for patients with comorbidities that are normally associated with elevated Hb levels (e.g., cyanotic heart disease).
| 3.7: | In all pediatric CKD patients receiving ESA therapy, we suggest that the selected Hb concentration be in the range of 11.0 to 12.0 g/dl (110 to 120 g/l). (2D) |
RATIONALE
As mentioned above, in children with CKD observational data associates high Hb with better survival139 and/or increased exercise capacity.142 Moreover, a recent North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) retrospective analysis done on pediatric CKD patients found an increased risk of hospitalization in children with low Hb compared to those with normal Hb.144 However, based on recent experience with the adult CKD patient population, caution is warranted with any extrapolation from observational treatment studies to conclusions on hard outcomes. This being said, direct extrapolation of the results from adult trials to pediatric patients is not appropriate given the differences in causes of CKD, contributions of age to growth and development, and impact of comorbidities on outcomes.
ESA DOSING
RATIONALE
The initiation of ESA therapy, ESA dose adjustments and rates of changes have remained similar to those outlined in the 2006 KDOQI Anemia Guideline.50 In general, the objective of initial ESA therapy is a rate of increase in Hb concentrations of 1.0 to 2.0 g/dl (10 to 20 g/l) per month. This is consistent with the findings in ESA trials of CKD-associated anemia where the mean initial rates of Hb concentration increase were of 0.7 to 2.5 g/dl (7 to 25 g/l) in the first 4 weeks. However, a rise in Hb of greater than 2.0 g/dl (20 g/l) over a 4-week period should be avoided.
The rate of increase varies greatly as a function of individual ESA responsiveness. Poor responders are more likely to be female, to have a history of cardiovascular disease (CVD), to have signs of iron deficiency and inflammation, and to be overweight.145 The response also depends on initial dose, dosing frequency, and route of administration. The dependence on dosing frequency and route of administration concerns epoetin-alfa, epoetin-beta, and darbepoetin but not CERA (continuous erythropoietin receptor activator [methoxy polyethylene glycol-epoetin-beta]). When ESAs were introduced into clinical practice over 20 years ago, hypertension was frequently noted in the first 3 months after initiating therapy in severely anemic patients, and seizures in rare instances. It is possible, although not proven, that these events were related to a too rapid rate of increase in Hb concentrations.
Epoetin-alfa or epoetin-beta dosing usually starts at 20 to 50 IU/kg body weight three times a week. Darbepoetin-alfa dosing usually starts at 0.45 µg/kg body weight once weekly by subcutaneous (SC) or IV administration, or 0.75 µg/kg body weight once every 2 weeks by SC administration. CERA dosing starts at 0.6 µg/kg body weight once every 2 weeks by SC or IV administration for CKD ND and CKD 5D patients, respectively, or 1.2 µg/kg body weight once every 4 weeks by SC administration for CKD ND patients. Higher baseline Hb concentrations require lower initial ESA doses, except for CERA for which there is no initial dose change. In patients with a history of CVD, thrombo-embolism or seizures, or in those with high blood pressure, the initial doses should be in the lower range. Epoetin-alfa or epoetin-beta dosage may subsequently be increased every 4 weeks by a weekly dose of 3 x 20 IU/kg if the increase of Hb is not adequate. Increases in dose should not be made more frequently than once a month. If the Hb is increasing and approaching 11.5 g/dl (115 g/l), the dose should be reduced by approximately 25%. If the Hb continues to increase, doses should be temporarily withheld until the Hb begins to decrease, at which point therapy should be reinitiated at a dose approximately 25% below the previous dose. Alternatively, one could simply repeat the Hb determination again in a shorter interval (e.g., weekly) and interpret any further rise, in particular in light of reticulocyte counts and their direction, before considering holding the dose. If the Hb increases by more than 1.0 g/dl (10 g/l) in any 2-week period, the dose should be decreased by approximately 25%. See Recommendations 3.13.1 to 3.15.2 regarding ESA hyporesponsiveness and loss of ESA response (Supplementary Table 20 online).
Dose adjustments may be necessary once the Hb target range has been reached. Note that in clinical practice, achieved Hb values may easily rise above or fall below the optimal Hb limits. Therefore, cautious dose adaptations are required. In general, ESA dose adjustments are made only after the first 4 weeks after ESA initiation. The frequency of ESA dose adjustment should be determined by the rate of increase in Hb concentrations during initial ESA therapy, the stability of Hb concentrations during maintenance ESA therapy, and the frequency of Hb testing. The minimum interval between ESA dose adjustments in the outpatient setting generally is 2 weeks because the effect of most dose changes will not be seen within a shorter interval. ESA doses should be decreased, but not necessarily held, when a downward adjustment of Hb concentration is needed. Withholding ESA doses, particularly for long periods, may lead to a delayed decrease in Hb concentrations to less than target range. Such a decrease may initiate periodic cycling of Hb concentrations at greater than and less than the target Hb range.146 Hb variability has been found to be an independent predictor of mortality in a large US CKD 5HD patient population147 although this observation could not be confirmed in a large European CKD 5HD patient cohort.148
Each time a patient with CKD is hospitalized the treating clinician should evaluate or reevaluate the patient's ESA requirements. Disease states such as severe infections or postsurgery may modify the ESA responsiveness profoundly. In case of profound anemia and markedly impaired ESA response a red cell transfusion may be preferred to administering ESAs or increasing ESA dose.
ESA ADMINISTRATION
RATIONALE
As outlined in the 2006 KDOQI guideline,50 the route of administration should be determined by the CKD stage, treatment setting, efficacy considerations, and the class of ESA used. Among CKD 5D patients undergoing intermittent hemodialysis or hemofiltration therapy, either SC or IV administration is possible. In the outpatient setting, SC administration is the only routinely feasible route of administration for patients with CKD 3-5 or on peritoneal dialysis treatment. Among short-acting ESAs, efficacy of SC administration in patients with CKD 5HD may be superior to that of IV administration, as shown by a large multicenter RCT in hemodialysis patients.149 However, another RCT of much smaller sample size did not find an advantage of SC over IV administration in CKD 5HD patients.150 Among long-acting ESAs, efficacy of SC compared with IV administration appears to be equivalent at examined dosing frequencies.151, 152, 153 Furthermore, CKD 5HD patients in general prefer IV to SC administration of ESAs because SC administration may be painful (Supplementary Tables 21-24 online).
Frequency of administration
| 3.10: | We suggest determining the frequency of ESA administration based on CKD stage, treatment setting, efficacy considerations, patient tolerance and preference, and type of ESA. (2C) |
RATIONALE
The frequency of ESA administration depends on considerations of efficacy, convenience and comfort. Maximum efficacy occurs within dosing intervals that are ESA class specific. For example, in patients on hemodialysis treatment receiving SC or IV short-acting ESA therapy, epoetin-alfa efficacy decreases when the dosing is extended from 3 times weekly to once-weekly administration,154 and even more so when the dosing intervals are extended to every other week administration.155 Among long-acting ESAs, darbepoetinalfa appears to have maximum efficacy when administered every 2 weeks, and methoxy polyethylene glycol-epoetin-beta (CERA) every 4 weeks.156 When converting short-acting ESAs to long-acting ESAs, differences in drug half-life need to be considered. For the sake of comparison, 3 times weekly administered epoetin-alfa to darbepoetin-alfa given only once monthly resulted in a decreased frequency of injections needed to maintain Hb concentrations of CKD patients within an accepted target range157 (Supplementary Tables 25-28 online).
When converting a patient from one ESA to another the pharmacokinetic and pharmacodynamic characteristics of the new ESA need to be taken into consideration. The manufacturers have provided conversions from epoetinalfa or epoetin-beta to darbepoetin-alfa or CERA. Note that the conversion ratios from epoetin to darbepoetin are non-linear.
When using different types of approved ESAs (biosimilars that have received approval by official regulatory bodies such as FDA and European Medicines Agency [EMA]), license information provided by companies should also be taken into account.
TYPE OF ESA
RATIONALE
As outlined above, the choice of short-acting or long-acting ESAs needs to take into account a number of different aspects, encompassing patient-oriented issues and country-specific considerations. At present, there is no evidence that any given ESA brand is superior to another in terms of patient outcomes, with the historical exception of the temporary increase in the incidence of antibody-mediated pure red cell aplasia (PRCA) about 10-20 years ago, which was associated with SC administration of an epoetin-alfa formulation available in Europe, but not in the United States.158, 159 It is the considered opinion of the Work Group that the likelihood of differences in clinical outcomes among ESA brands is low, although there is no robust evidence supporting this assumption (Supplementary Tables 29-32 online).
At present, a number of different types of short-acting or long-acting ESAs are available worldwide, including original formulations, biosimilars, and 'copy' ESAs which have not been exposed to the rigor of scientific evaluation as mandated by the regulatory agencies prior to approval. Their accessibility and costs vary from country to country. True biosimilars, as defined by the EMA, are not identical to the originator products, but they have undergone a minimum number of regulatory 'equivalence' or 'non-inferiority' studies to gain marketing authorization in Europe. In other countries outside Europe, some 'copy' ESA products have been marketed that may not have undergone the same rigorous testing.160 Since patient safety is one of the most important drug treatment issues, only biosimilars approved by an independent regulatory agency should be used.
EVALUATING AND CORRECTING PERSISTENT FAILURE TO REACH OR MAINTAIN INTENDED HEMOGLOBIN CONCENTRATION
Frequency of monitoring
ESA initiation phase. The suggestion to monitor Hb values at least monthly in patients in whom ESA therapy is started is intended to provide sufficient surveillance information to assist in achieving and maintaining desired Hb concentrations safely and follows common practice.50 The minimum interval between ESA dose adjustments is 2 weeks because the effect of most dose changes will not be seen within a shorter interval. Consideration of an ESA dose adjustment is based on the next projected Hb concentration. Because the accuracy of projection (extrapolation) increases with the number of contributing data points, the frequency of Hb monitoring is likely to be an important determinant of the accuracy of ESA dose adjustment. However, evidence to support this line of reasoning is indirect. Several RCTs have randomized CKD 5HD patients with target-range Hb concentrations to a change in frequency of ESA administration, a change in ESA class, or both. RCTs that have monitored Hb values weekly and adjusted ESA doses as frequently as every 2 weeks have achieved stable Hb concentrations early after randomization.152, 161, 162 In contrast, an RCT that monitored Hb concentrations and considered ESA dose adjustment monthly required 6 to 9 months to stabilize Hb concentrations after randomization,163 but mean Hb concentration remained within the target range for that trial.
ESA maintenance phase. Within the recommended ranges for monitoring and dose adjustment, unstable Hb concentration, inappropriate high or low Hb concentration, and hemodialysis favor shorter intervals of ESA administration, whereas stable Hb concentration, within target Hb concentration, peritoneal dialysis, CKD 3-5, and minimizing laboratory resource utilization favor longer intervals for long-acting ESAs such as darbepoetin. The frequency of ESA dose adjustment is unaffected by length of action: during an 8-week period with weekly Hb monitoring, about equal numbers of patients receiving either short-acting ESA thrice weekly or darbepoetin once weekly required dose adjustments (44% and 49%, respectively).162
Initial ESA hyporesponsiveness
Subsequent ESA hyporesponsiveness
Management of poor ESA responsiveness
RATIONALE
Relative resistance to the effect of ESAs is a common problem in managing the anemia of patients with CKD and remains the subject of intense interest, all the more since ESA hyporesponsiveness has been found to be among the most powerful predictors of the risk of cardiovascular events and mortality.164 Recently a report from TREAT assessed the initial Hb response to darbepoetin after two weight-based doses at 2 weekly intervals, in 1872 patients with CKD and diabetes.145 Patients with a poor response, (the lowest quartile, who had o2% change in Hb concentration after 1 month), had higher rates of the composite cardiovascular events (adjusted HR 1.31, 95% CI 1.09-1.59), compared to those with a better response. Although this differential effect may be related to comorbidity in hyporesponsive patients, nonetheless it is possible that the high ESA doses used in hyporesponsive patients may be toxic. Though not empirically tested, per se, the definition of initial hyporesponsiveness agreed upon by the Work Group is derived from the secondary analysis of the TREAT study.145 Since a <2% increase in the Hb concentration is likely to be within the variability range of Hb values in individual patients, this value is considered as "no increase." The definition of initial hyporesponsiveness relies on presently accepted ESA starting doses, as indicated in the Rationale under 3.8.1-3.8.4. Of note, weight-based doses for darbepoetin do not differ for IV or SC routes, but do differ for epoetin-alfa.
If lower initial dosages than those used in TREAT are chosen, the diagnosis of hyporesponsiveness must take this into account. For example, in the USA the label for darbepoetin now recommends a starting dose of 0.45 µg per kg per four weeks, much lower than the dose used in TREAT or in Europe (i.e., 0.45 µg per kg per week or 0.75 µg per kg per two weeks). If such lower starting doses are used, repeated escalations in ESA dose should be allowed to reach double the weight-based dose used in TREAT.
Although the distinction between initial ESA hyporesponsiveness and acquired partial or complete loss of ESA responsiveness in a patient with already treated, stable anemia is somewhat artificial, it is useful in our opinion for clinical practice.
In the Normal Hematocrit Study both the high Hb and the low Hb groups revealed an inverse relationship between achieved Hb and the primary outcome (death or myocardial infarction).118 This is consistent with the idea that those patients who failed to achieve the target Hb were unable to do so because comorbid condition(s) existed that prevented achievement of this target. Thus, hyporesponsiveness may just have been a marker for adverse outcomes, although the possibility that high ESA doses used in hyporesponsive patients are toxic in themselves cannot be excluded. Dose targeting bias has been reported by the Kidney Disease Clinical Studies Initiative Hemodialysis Study (HEMO) investigators.165 In this RCT ESRD patients, randomly allocated to either high or low quantity of dialysis, as measured by Kt/V, demonstrated an inverse relationship between achieved Kt/V and mortality. The interpretation was that patients with comorbid conditions were unable to achieve higher Kt/V and that comorbidity predisposed these patients to earlier death.
The same principle as used with defining hyporesponsiveness to darbepoetin could be applied to the early response to other short-acting ESAs but cannot be applied to longer acting ESAs such as CERA. In that case, evaluating the Hb response after a time period of 2 months appears to be appropriate. Early ESA hyporesponsiveness or the subsequent occurrence of hyporesponsiveness in CKD patients with previously stable Hb values should lead to an intensive search for potentially correctable factors which might be causally involved. Unfortunately, besides iron deficiency, there are only few other easily reversible factors that contribute to ESA hyporesponsiveness, as shown in Table 3. If other such factors are identified they should be treated as well. Although most disorders associated with hyporesponsiveness are readily apparent, hyporesponsive patients should be evaluated for coexisting oncological or hematologic disorders. They include hematological and non-hematological malignancies as well as such diverse hematological conditions as thalassemia, sickle cell disease or the anemia associated with other chronic diseases. Myelodysplastic syndromes are a particular case. If at all ESA responsive, the anemia in patients with myelodysplastic syndrome responds more slowly. Therefore, 1 month may be too short to define hyporesponsiveness in this and several other conditions. Moreover, patients with myelodysplastic syndromes may need higher ESA doses. Finally, a rare disorder, PRCA, deserves special consideration (see 3.17.1-3.17.3). The estimation of loss of ESA response also may require a longer observation time in some patients. Note that poor ESA response, either in the initial correction phase or subsequently, is most often a transient condition. Complete loss of response is exceptional. Poor responders should periodically be re-tested for responsiveness, including after the correction of treatable causes of hyporesponsiveness.
Table 3 | Potentially correctable versus non correctable factors involved in the anemia of CKD, in addition to ESA deficiency
| Easily correctable | Potentially correctable | Impossible to correct |
|---|---|---|
|
Absolute iron deficiency Vitamin B12/folate deficiency Hypothyroidism ACEi/ARB Non-adherence |
Infection/inflammation Underdialysis Hemolysis Bleeding Hyperparathyroidism PRCA Malignancy Malnutrition |
Hemoglobinopathies Bone marrow disorders |
ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin-receptor blocker;
PRCA, pure red cell aplasia.
It is important to note that the dosing requirements may differ substantially between children and adults. Registry data from NAPRTCS showed that young children require higher doses of ESA than adults, ranging from 275 U/kg/week to 350 U/kg/week for infants and 200-250 U/kg/week for older children.166 Another retrospective analysis among patients on chronic hemodialysis found that children and adolescents required higher absolute doses of ESA than adults to maintain target hemoglobin levels, despite the lower mean body weight of the children.167 Unfortunately, there are no RCTs that establish the appropriate dosing of ESA in children. Future research to establish pediatric ESA dosing guidelines is needed, especially for infants and younger children.
There may be toxicity from high doses of ESA, as suggested, though not proven, by recent post-hoc analyses of major ESA RCTs,145, 168 especially in conjunction with the achievement of high Hb levels.169 Therefore, in general ESA dose escalation should be avoided. The Work Group suggestions for initial and acquired hyopresponsiveness imply that maximal doses should be no greater than four times initial weight-based appropriate doses.
Table 4 | Practical approach in presence of ESA hyporesponsiveness
| Tests | Finding and action |
|---|---|
| 1. Check adherence | If poor, attempt to improve (if self-injection) |
| 2. Reticulocyte count | If >130,000/µl, look for blood loss or hemolysis: endoscopy, colonoscopy, hemolysis screen |
| Serum vitamin B12, folate | If low, replenish |
| Iron status | If low, replenish iron |
| Serum PTH | If elevated, manage hyperparathyroidism |
| Serum CRP | If elevated, check for and treat infection or inflammation |
| Underdialysis | If underdialyzed, improve dialysis efficiency |
| ACEi/ARB use | If yes, consider reducing dose or discontinuing drug |
| 3. Bone marrow biopsy | Manage condition diagnosed e.g., dyscrasia, infiltration, fibrosis |
ACEi, angiotensin-converting enzyme inhibitor; ARB, angiotensin-receptor blocker; CRP, C-reactive protein; PTH, parathyroid hormone.
In practice, Tables 3 and 4 can guide to diagnose and correct ESA hyporesponsiveness. In patients in whom all correctable causes have been maximally treated but who remain hyporesponsive, ESA therapy may be continued cautiously at doses up to 4 times the initial dose to prevent a further decline in Hb concentration. Red cell transfusions can be used to prevent or treat anemia-related symptoms and signs. The treatment strategy needs to take into account each patient's anemia tolerance and potential benefits and risks linked to increases in Hb values solely obtained by high ESA dosing.
Given the disproportionate burden of morbidity and mortality that the hyporesponsive patient population bears and the ESA expense that hyporesponsiveness engenders, further research is necessary on the causes and management of hyporesponsiveness.
ADJUVANT THERAPIES
RATIONALE
Several adjuvant treatments have been proposed, either with the goal of limiting the use of more expensive ESA therapy or to improve ESA responsiveness.
Androgens. The use of androgens for treatment of anemia was suggested long before rHuEPO became available in clinical practice. Androgens were used regularly in many centers in the treatment of anemia in dialysis patients despite the need for intramuscular (IM) injection and a variety of adverse events, including acne, virilization, priapism, liver dysfunction, injection-site pain, and risk for peliosis hepatis and hepatocellular carcinoma. The three RCTs that tested androgens in combination with ESA therapy in CKD 5HD patients were all small short-term studies. Currently recommended Hb concentrations were not achieved, and in two of them the ESA doses used were lower than current practice.170, 171, 172 The studies did not enroll patients with ESA hyporesponsiveness, so the effect of androgens on hyporesponsiveness is unknown. The risks of androgen therapy and their uncertain benefit on Hb concentration or clinical outcomes argue against their use as an ESA adjuvant.
Vitamin C. Vitamin C has been reported to increase the release of iron from ferritin and the reticuloendothelial system and increase iron utilization during heme synthesis. 173, 174 A recent meta-analysis of vitamin C use in CKD 5HD175 and a more recent small RCT176 concluded that vitamin C may result in larger increases in Hb and may limit the use of ESAs. In seven trials, patients generally had functional iron deficiency and in three studies they had EPO hyporesponsiveness (variously defined).176, 177, 178 However, the number of patients studied was insufficient to address the safety of this intervention. Thus the long-term safety of IV ascorbic acid in HD patients remains undefined, and whether secondary oxalosis should be a concern.
Convincing data do not exist for other potential adjuvants including vitamin D, vitamin E, folic acid, L-carnitine and pentoxifylline. Several anecdotal reports, small case series, and nonrandomized studies, primarily in CKD 5HD patients, have been published, but do not provide sufficient evidence upon which to base a recommendation. Future RCTs are clearly needed for ESA adjuvants.
EVALUATION FOR PURE RED CELL APLASIA (PRCA)
RATIONALE
Rarely, patients undergoing ESA therapy develop antibodies that neutralize both ESA and endogenous erythropoietin. The resulting syndrome, antibody-mediated PRCA, is characterized by the sudden development of severe transfusion- dependent anemia. Rapid recognition, appropriate evaluation, and prompt intervention can be effective in limiting the consequences of this life-threatening condition. Antibody-mediated PRCA, although rare in patients administered ESAs, received urgent attention after 1998. Between 1989 and 1998, three reports described the development of PRCA in only a small number of patients with CKD administered ESAs. Reports of PRCA increased sharply in 1998 and reached a peak in 2002.159, 179 These reports were associated with SC administration of an epoetin-alfa formulation not available in the United States. After removal of this formulation from the market, by 2004, the incidence of new antibody-mediated PRCA had decreased to pre-1998 levels. Isolated cases of PRCA have been observed in association with the use of other ESAs.159, 179, 180 Outside this historical episode the incidence rate of PRCA with SC use of all other forms of SC-administered ESA is estimated to be 0.5 cases/10,000 patient-years.158 Antibody-associated PRCA stemming from IV administration of ESAs is rare and has only been reported anecdotally.181
Recommendations based on expert opinions have been published to guide the workup and therapy of patients suspected to have antibody-mediated PRCA.179, 182, 183, 184 The two main distinguishing features of antibody-mediated PRCA are the associated decline in blood Hb concentration of approximately 4 g/dl (40 g/l) per month, and a decrease in the number of circulating reticulocytes to <10,000/µl of blood.185 Bone marrow biopsy characteristically shows reduced numbers or absence of erythroblasts. The definitive diagnosis is dependent upon demonstration of the presence of neutralizing antibodies against erythropoietin. Evidence for parvovirus infection as an alternative cause of PRCA should be sought and excluded.
Following a diagnosis of antibody-mediated PRCA, patients should stop treatment with the incriminated ESA immediately and not resume treatment with the same or another EPO-derived ESA.184 Immunosuppressive therapy may hasten the disappearance of circulating antibodies in patients with EPO-induced PRCA, and allow endogenous erythropoiesis to recover to pre-treatment levels. In a retrospective study of 47 patients who developed PRCA during EPO therapy (primarily epoetin brand 'Eprex®' in Europe), 29 of 37 patients (78%) who received immunosuppressive therapy recovered, whereas none of the nine patients who did not receive immunosuppressive therapy recovered.185 Red cell production recovered only when patients received immunosuppressive treatment. Re-exposure to epoetins or darbepoetin-alfa can re-induce the formation of antibodies.186 Anaphylactoid reactions after repeated injections of epoetin- or darbepoetin-alfa have been reported in a patient with pure red-cell aplasia.187 A novel approach to the treatment of this condition using a synthetic, peptide-based erythropoietin-receptor agonist (peginesatide) has generated optimistic results,188 and has the advantage of avoiding immunosuppressive therapy.
The recognition of antibody-mediated PRCA in patients treated with recombinant epoetins has underscored the need for full clinical documentation and post-marketing surveillance with newer ESAs and biosimilar products, as well as therapeutic recombinant proteins in general.189
If a decision to treat with peginesatide is taken, it can be initiated at a dose of 0.05 to 0.075 mg/kg body weight by subcutaneous injection every 4 weeks. Subsequently, the dose needs to be adjusted to reach the desired target Hb value.
RESEARCH RECOMMENDATIONS
The following research questions have arisen during the deliberations of the Work Group, and further research will be necessary to answer them.
- In cohort studies moderate anemia is associated with an increased incidence of cardiovascular events. Is anemia really a risk factor for these events or is it a marker for some other cardiovascular risk factor(s)?
- There is uncertainty about optimal Hb targets for ESA therapy. What is the risk-benefit ratio of low Hb targets <10.0 g/dl (<100 g/l) or high targets of 11.5-13.0 g/dl (115-130 g/l), compared to conventional targets of 10.0-11.5 g/dl (100-115 g/l)?
- These guidelines have stressed individualization of anemia therapy. Should the objective of anemia therapy be improvement in clinical outcomes (provided Hb concentration is <13.0 g/dl [<130 g/l]) rather than achievement of a specified Hb target range? Should these outcomes include improvements in QoL, and if so, what defines clinically important improvements?
- As the relationship between ESA responsiveness and hard patient outcomes may be the result of co-morbidity or of high ESA dose, what is the impact of high vs low dose on clinical outcomes in ESA hyporesponsive patients?
- Is the risk-benefit ratio of anemia correction similar in non-diabetic and diabetic CKD patients?
- Is there a difference in adverse clinical outcomes comparing IV and SC routes of administration?
- Are the risk-benefit ratios for biosimilars comparable to current ESAs?
- What is the pathogenesis of cerebrovascular and vascular toxicity associated with normalization of Hb using ESAs?
- Are CKD patients with cancer or a cancer history who are receiving ESA therapy at higher cardiovascular risk than non-CKD patients with cancer or a cancer history?
- What is the effect of vitamin C administration in functional iron deficiency and what is the clinical impact of increased oxalate levels?
- There appears to be differences in anemia treatment outcomes between different geographic regions. What are the reasons for this?
- What are the risks and benefits of ESA administration on outcomes in anemic children with CKD?
- What are the appropriate, weight-based, dosing regimens for the younger pediatric patients, especially those under the age of two years?
DISCLAIMER
While every effort is made by the publishers, editorial board, and ISN to see that no inaccurate or misleading data, opinion or statement appears in this Journal, they wish to make it clear that the data and opinions appearing in the articles and advertisements herein are the responsibility of the contributor, copyright holder, or advertiser concerned. Accordingly, the publishers and the ISN, the editorial board and their respective employers, office and agents accept no liability whatsoever for the consequences of any such inaccurate or misleading data, opinion or statement.While every effort is made to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this Journal, should only be followed in conjunction with the drug manufacturer's own published literature.
SUPPLEMENTARY MATERIAL
Supplemental Table 7: Association between anemia severity (prior to erythropoietin use) and clinical outcome in multivariable analyses.
Supplemental Table 8: Association between hyperparathyroidism and ESA responsiveness in multivariable analyses.
Supplemental Table 9: Evidence profile of RCTs comparing higher vs. lower Hb targets/ESA doses in the HD-CKD and PD-CKD populations.
Supplemental Table 10: Summary table of RCTs comparing different Hb targets/ESA doses on key clinical outcomes in the HD-CKD and PD-CKD populations.
Supplemental Table 11: Summary table of RCTs comparing different Hb targets/ESA doses on quality of life in the HD-CKD and PD-CKD populations.
Supplemental Table 12: Summary table of RCTs comparing different Hb targets/ESA doses on Fatigue, Vitality/Energy, and Physical function in the HD-CKD and PD-CKD populations.
Supplemental Table 13: Summary table of RCTs comparing different Hb targets/ESA doses on non-CVD/mortality adverse event rates in the HD-CKD and PD-CKD populations.
Supplemental Table 14: Summary table of RCTs comparing different Hb targets/ESA doses on exercise capacity in the HD-CKD and PD-CKD populations.
Supplemental Table 15: Evidence profile of RCTs comparing different higher vs. lower Hb targets/ESA doses in the ND-CKD populations.
Supplemental Table 16: Summary table of RCTs comparing different Hb targets/ESA doses on key clinical outcomes in the ND-CKD population.
Supplemental Table 17: Summary table of RCTs comparing different Hb targets/ESA doses on quality of life in the ND-CKD population.
Supplemental Table 18: Summary table of RCTs comparing different Hb targets/ESA doses on Fatigue, Vitality/Energy, and Physical function in the ND-CKD population.
Supplemental Table 19: Summary table of RCTs comparing different Hb targets/ESA doses on non-CVD/mortality adverse event rates in the ND-CKD population.
Supplemental Table 20: ESA protocols from the major trials in CKD populations.
Supplemental Table 21: Evidence profile of RCTs examining IV vs. SC EPO in CKD patients with anemia.
Supplemental Table 22: Summary table of RCTs examining IV vs. SC ESA in CKD patients with anemia (categorical outcomes).
Supplemental Table 23: Summary table of RCTs examining IV vs. SC ESA in CKD patients with anemia (continuous outcomes).
Supplemental Table 24: Summary table of adverse events in RCTs examining IV vs. SC EPO in CKD patients with anemia.
Supplemental Table 25: Evidence profile of RCTs examining different dosing schedules in CKD patients with anemia.
Supplemental Table 26: Summary table of RCTs examining different dosing schedules in CKD patients with anemia (categorical outcomes).
Supplemental Table 27: Summary table of RCTs examining different dosing schedules in CKD patients with anemia (continuous outcomes).
Supplemental Table 28: Summary table of adverse events in RCTs examining different dosing schedules in CKD patients with anemia.
Supplemental Table 29: Evidence profile of RCTs examining ESA vs. ESA in CKD patients with anemia.
Supplemental Table 30: Summary table of RCTs examining ESA vs. ESA in CKD patients with anemia (categorical outcomes).
Supplemental Table 31: Summary table of RCTs examining ESA vs. ESA in CKD patients with anemia (continuous outcomes).
Supplemental Table 32: Summary table of adverse events in RCTs examining ESA vs. ESA in CKD patients with anemia (categorical outcomes).
Supplementary material is linked to the online version of the paper at http://www.kdigo.org/clinical_practice_guidelines/anemia.php
Chapter 4: Red cell transfusion to treat anemia in CKD
USE OF RED CELL TRANSFUSION IN CHRONIC ANEMIA
Repeated transfusions or use of an erythropoiesis-stimulating agent (ESA) are treatment options for chronic anemia in CKD. The choice between these depends on their relative benefits and harms, which vary among patients. For example, patients with a previous stroke have the greatest absolute risk of ESA-related stroke,127 whereas multiparous women have the highest risk of allosensitization with transfusion.190, 191 Although the clinical importance of allosensitization is disputed, it may delay or reduce the possibility of future kidney transplantation.
RATIONALE
As with any treatment, the use of red cell transfusions should be considered in terms of the balance of benefit and harms. The primary benefit is in maintaining sufficient oxygencarrying capacity and improvement in anemia-related symptoms.192 The harms are summarized in Tables 5 and 6 and discussed further below. This balance must also be considered alongside the balance between the benefits and harms of ESA therapy which is an alternative treatment for the anemia of CKD. The benefits and harms of ESA therapy are discussed in detail in Chapter 3, but, in summary, the benefits include improvement in anemia-related symptoms and reduced need for transfusion, and the most important harms are increased risk of stroke, thromboembolic events, and cancer progression or recurrence. When choosing between these two treatments for anemia in an individual, patient characteristics which influence the balance between benefits and harms for each treatment should be considered. These include history of stroke and previous or current cancer which place patients receiving ESA therapy at much higher absolute risk of these two problems. Conversely, patients potentially eligible for kidney transplantation have the greatest potential harm from transfusion, in terms of allosensitization,191, 193, 194 although the clinical importance of allosensitization is disputed. Previously transplanted patients and multiparous women seem to have the greatest absolute risk of allosensitization.190, 191
A related issue is when should the decision to treat a patient with either an ESA or a transfusion be made? This decision is subtly different for the two types of treatment as ESAs may be used to avoid transfusion and therefore before the need for transfusion has arisen i.e., in a prophylactic sense. Furthermore, the magnitude of the potential harms of transfusion (e.g., from infection) and some of the benefits from ESAs (e.g., transfusion avoidance) is dependent on the threshold for transfusion. If that threshold is high (i.e., transfusion is reserved until symptoms become severe or the Hb reaches a very low level) the risks related to transfusion will be low and the benefit of ESA therapy in avoiding transfusions will be small. Unfortunately, there is no consensus about when transfusion is indicated although we do know that the rate of transfusion increases markedly when the Hb falls below 10 g/dl (100 g/l);122, 127 whether that simply reflects practice-patterns or represents clear clinical need is uncertain. The following trials give examples of transfusion rates in CKD 5D and CKD ND patients. The trial conducted by the Canadian Erythropoietin Study Group, published in 1990, enrolled 118 CKD 5HD patients Hb <9.0 g/dl (<90 g/l), 49 (42%) of whom were described as 'transfusion-dependent'.122 The patients averaged approximately 7 transfusions each in the previous 12 months. These patients were randomized, equally, to 6 months treatment with placebo, erythropoietin with a target Hb 9.5-11.0 g/dl (95-110 g/l), or erythropoietin with a target Hb 11.5-13.0 g/dl (115-130 g/l). After 8 weeks, 23 patients in the placebo group received a blood-transfusion, compared with one in each of the two erythropoietin groups (for a gastrointestinal hemorrhage and following surgery). More recently, in the Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT), published in 2009, 4038 patients with diabetes, CKD ND and anemia (Hb≤11.0 g/dl [≤110 g/l]), were randomized, equally, to darbepoetin-alfa with target Hb 13 g/dl (130 g/l) or to placebo, with 'rescue' darbepoetin-alfa when Hb fell below 9.0 g/dl (90 g/l).127 Over a median followup of 29 months, 297/2012 (15%) patients randomized to darbepoetin-alfa and 496/2026 (25%) assigned to placebo received red cell transfusions (HR 0.56, 95% CI 0.49-0.65, P<0.001).
Table 5 | Estimated risk associated with blood transfusions per unit transfused
| Adverse event | Estimated risk* |
|---|---|
| Immunological | |
| Fever/allergic reactions | 1 in 100-200a,b |
| Hemolytic reaction | 1 in 6000b |
| Transfusion-related acute lung injury (TRALI) | 1 in 12,350a |
| Anaphylaxis | 1 in 50,000b |
| Fatal hemolysis | 1 in 1,250,000a |
| Graft versus host disease (GVHD) | Rare |
| Other | |
| Mistransfusion | 1 in 14,000-19,000c |
*United States data.
aData from Carson JL et al.212
bData from Klein.213
cData from Klein HG et al.214
We suggest that the decision to transfuse in the patient with non-acute anemia related to CKD should not be based upon any arbitrary Hb threshold and should, instead, be determined by the occurrence of symptoms and signs caused by anemia. We recognize that symptoms such as dyspnea and fatigue are non-specific, and that anemia-related symptoms may occur at different Hb levels in different patients.
Risks of blood transfusion
Risks associated with blood transfusion include transfusion errors, volume overload, hyperkalemia, citrate toxicity (leading to metabolic alkalosis and hypocalcemia), hypothermia, coagulopathy, immunologically-mediated transfusion reactions, including transfusion-related acute lung injury (TRALI), and iron overload, all of which are uncommon (Table 5).190, 195, 196, 197, 198, 199, 200, 201, 202, 203, 204, 205, 206, 207 Transmission of infections, although rare, is a major concern and this risk varies between countries (Table 6).208, 209, 210, 211 These complications are reviewed extensively elsewhere. The importance of human leukocyte antigen (HLA) sensitization is disputed and discussed in more detail below.
HLA sensitization. The risk of sensitization after blood transfusion has changed over time probably, at least in part, due to changes in blood transfusion practices and the use of more precise methods to measure allosensitization.
In the early 1980s, Opelz et al. examined the risk of sensitization in 737 CKD 5HD patients (Figures 3A and 3B), of whom 331 were followed prospectively (Figure 3C).190 Approximately 90% of all transfusions were given in the form of 'packed cells' and antibodies were measured by the lymphocyte cytotoxicity test. Overall, 28% of patients followed prospectively developed HLA antibodies. Of these, 18% developed reactivity to 10-50% of the panel, 7% to 50-90%, and <3% to >90% of the panel after up to 20 transfusions (Figure 3C). Among men, 90% remained 'unresponsive' (<10% antibody reactivity against the panel) and 10% developed reactivity to 10-50% of the panel (Figure 3C). In contrast, after 10 transfusions, only 60% of the women were 'unresponsive,' 11% demonstrated 10-50% reactivity, 23% 51-90% reactivity, and 6% >90% reactivity (Figure 3C). These data suggested that the main drivers of HLA sensitization following red cell transfusion are previous pregnancies and previous transplantation. The data also suggested that men have a much lower risk of HLA sensitization following transfusion than women, and women with multiple pregnancies have a much greater risk of HLA sensitization than nulliparous women. However, more recent data from the US Renal Data System (USRDS) 2010 Annual Report,191 have challenged this assumption, suggesting that males receiving previous blood transfusions may also be at increased risk.
Table 6 | Estimated risk of transfusion-related infections per unit transfused
| Potential transfusion-related risks | Estimated risk* |
|---|---|
| Hepatitis B | 1 in 282,000-1 in 357,000a |
| West Nile virus | 1 in 350,000b |
| Death from bacterial sepsis | 1 in 1,000,000b |
| Hepatitis C | 1 in 1,149,000a |
| Human immunodeficiency virus (HIV) | 1 in 1,467,000a |
*United States data.
aData from Carson JL et al.212
bData from Rawn J.215
Studies performed in the last two decades showed that the risk of sensitization with blood transfusion is apparently lower than previously reported, with an overall response rate ranging from 2 to 21%.216, 217, 218 A possible, albeit controversial, explanation for this lower sensitization rate is that red cell transfusions in recent years are less immunogenic because they contain fewer leukocytes due to widespread use of blood filters.
Other tentative conclusions from previous studies include the following: a) washed-red cells do not appear to be less immunogenic than non-washed red cells;190 b) no consistent reduction in sensitization has been demonstrated with donor-specific217 and HLA-DR matched transfusions;219 c) higher numbers of blood transfusions have been associated with an increased risk of sensitization in some studies220, 221 but not in others.190, 222
However, more recent data from the USRDS indicates that risk of sensitization with blood transfusions is substantial. For example, compared with patients who have never received a blood transfusion, patients who received transfusions have an odds ratio of having panel reactive antibody (PRA)>80% of 2.38.191 Interestingly, in this analysis the risk of being highly sensitized at the time of transplantation was higher for men than for women.
Figure 3 | Lymphocytotoxic antibody reactivity against random donor test panel in relation to the number of blood transfusions. Fractions of patients reacting against <10%, 10 to 50%, 51 to 90% and >90% of the panel donors are plotted. All 737 patients were on chronic hemodialysis, waiting for a first kidney transplant. Numbers of patients after 2, 5, 10, 15, and 20 transfusions are indicated at top of graphs. (A) Male and female patients. (B) Females patients separated by the number of previous pregnancies. (C) Lymphocytotoxic antibodies in patients who were studied prospectively throughout the course of treatment. Reprinted from Opelz G, Graver B, Mickey MR et al. Lymphocytotoxic antibody responses to transfusions in potential kidney transplant recipients. Transplantation 1981; 32(3): 177-183 (ref. 190) with permission from Lippincott Williams & Wilkins; accessed http://journals.lww.com/transplantjournal/Abstract/1981/09000/Lymphocytoxic_Antibody_Responses_to_Transfusions.2.aspx
Effect of leukocyte-reduced blood transfusions on sensitization. Although, leukocytes may be a contributor to, if not the cause of, a number of adverse consequences of blood transfusion, including immunologically-mediated effects, infectious disease transmission, and reperfusion injury, leukoreduction of blood products does not decrease sensitization in previously transplanted or in potential future kidney transplant candidates.223, 224, 225 One recent study reported that male patients awaiting their first organ transplant had a fourfold increased risk of developing HLA antibody if they had been previously transfused when compared with those who did not have a history of a transfusion.226 Thus, transfusion in the post-leukodepletion era still continues to pose a significant risk of sensitization. A possible reason for this finding is that the number of HLA molecules contributed by the red cells is comparable to that of leukocytes.227
Table 7 | Indications for blood transfusions
| Indication | Comments |
|---|---|
| When rapid correction of anemia is required to stabilize the patient's condition (e.g., acute hemorrhage, unstable myocardial ischemia) |
|
| When rapid pre-operative Hb correction is required |
|
| When symptoms and signs related to anemia are present in patients in whom ESA therapy is ineffective (e.g., bone marrow failure, hemoglobinopathies, ESA resistance) |
|
| When symptoms and signs related to anemia are present in patients in whom the risks of ESA therapy may outweigh the benefits |
|
CKD, chronic kidney disease; ESA, erythropoiesis-stimulating agent; Hb, hemoglobin.
Association between sensitization and delay in organ transplantation. According to USRDS data reported in 2010, the mean wait-time to transplant for patients listed between 1991 and 2008 was an average of 2 months longer for transfused than non-transfused patients in the United States.191 Increased PRA titers, whether due to blood transfusions or other factors, were associated with a longer wait to find a compatible donor and may have completely precluded transplantation in some patients. Non-sensitized patients (0% PRA at the time of listing) had the shortest wait-time (median of 2.5 years in 2005) while those with a PRA of 1-19% and 20-79% had median wait-times of 2.9 and 4.3 years, respectively. Highly sensitized patients (≥80% PRA) waited the longest and in these patients a median wait-time could not be calculated for patients listed in 2005. As a result of the delay in finding compatible donors in patients with PRA ≥80%, the percentage of these patients increased from 7.5% at listing to 13.3% five years after listing.
Figure 4 | Algorithms for red cell transfusion use in CKD patients. ESA, erythropoiesis-stimulating agent; Hb, hemoglobin.
Not being transplanted, or having to wait longer for transplantation, is associated with lower survival.228, 229 Receiving a transfusion while on the transplant wait list is associated with a nearly 5-fold higher risk of dying while on the wait list in the first five years, and an 11% reduction in the likelihood of receiving a transplant within the first five years.191, 230 In transplanted patients, the presence of preformed HLA antibodies is associated with an increased risk of early and late graft loss.193, 194, 231, 232 Recent data also suggest that pre-existing donor-specific HLA antibodies identified by a Luminex single-antigen assay at the time of transplantation are associated with a higher incidence of antibody-mediated rejection and inferior graft survival.233
URGENT TREATMENT OF ANEMIA
RATIONALE
In certain urgent clinical situations, red cell transfusion may be needed for the immediate correction of anemia. These include acute severe hemorrhage and other clinical problems caused by, or exacerbated by, anemia, such as acute myocardial ischemia. When urgent surgery is required, transfusion may also be given to achieve rapid preoperative correction of Hb. The Hb threshold for transfusion in this situation is uncertain but we suggest that this treatment be considered if the Hb is <7 g/dl (<70 g/l).
Table 7 and Figure 4 summarize the approaches to the use of red cell transfusions in patients with CKD.
RESEARCH RECOMMENDATIONS
There is a lack of randomized controlled trials on the use of blood transfusions as a primary intervention in patients with anemia and CKD. Given the logistical difficulties in conducting such trials, it is likely that observational data will continue to predominate in this therapeutic area.
Future research should include:
- Prospective observational data collection on the use of red cell transfusions in CKD patients, particularly dialysis patients, including the reason(s) for transfusion, intent to list for future kidney transplantation, likelihood of receiving a kidney transplant, and graft outcomes.
- Prospective observational evaluation of the impact of red cell transfusions on the level of HLA sensitization.
- Given a striking disparity in the use of blood transfusions between the US and Europe, Canada and Australia in the TREAT study, and between in the US and Europe in the Phase 3 peginesatide clinical trial program, further research is needed to ascertain the 'drivers' for transfusion in CKD patients. Is this related to practice patterns or a real higher clinical need for transfusions in the US?
DISCLAIMER
While every effort is made by the publishers, editorial board, and ISN to see that no inaccurate or misleading data, opinion or statement appears in this Journal, they wish to make it clear that the data and opinions appearing in the articles and advertisements herein are the responsibility of the contributor, copyright holder, or advertiser concerned. Accordingly, the publishers and the ISN, the editorial board and their respective employers, office and agents accept no liability whatsoever for the consequences of any such inaccurate or misleading data, opinion or statement. While every effort is made to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this Journal, should only be followed in conjunction with the drug manufacturer's own published literature.